
Abstract
Background:
Aberrant expression of oncogenes and/or tumor suppressor genes (TSGs) drives the tumorigenesis and development of thyroid cancer. We investigated the expression and function of a member of the activating transcription factor (ATF)/cAMP-responsive element-binding protein (CREB) transcription factor (TF) family, ATF3, in thyroid cancer.
Methods:
Data from 80 patients with papillary thyroid cancer (PTC) in the First Affiliated Hospital of Sun Yat-sen University and 510 PTC samples in The Cancer Genome Atlas thyroid cancer database were utilized for gene expression and prognosis analyses. The survival data were analyzed by Kaplan–Meier curves and Cox regression with adjustment for age, sex, multilocality, extrathyroidal extension, lymph metastases, and history of neoadjuvant treatment. DNA methylation was analyzed by methylation-specific polymerase chain reaction (PCR) and bisulfite sequencing PCR. TFs binding to ATF3 promoter were identified by DNA pull-down combined with mass spectrum assay, and confirmed by quantitative PCR (qPCR), luciferase reporter assay, and chromatin immunoprecipitation (ChIP)-qPCR. We conducted functional assays in vitro and in a xenograft mouse model to evaluate the function of ATF3 in thyroid cancer. Integrated analyses based on RNA sequencing, ChIP-seq, and CUT&Tag assays were performed to explore the mechanisms underlying the function of ATF3.
Results:
ATF3 was significantly downregulated in PTC and patients with low ATF3 expression had reduced progression-free survival (adjusted hazard ratio = 0.50 [CI 0.26–0.98], p = 0.043). DNA hypermethylation in ATF3 promoter disrupted the binding of SP1 and MYC-MAX, leading to inactivation of the gene. ATF3 functioned as a TSG by inhibiting the proliferation and mobility of thyroid cancer cells. And ATF3 regulated the expression of a number of genes by binding to the regulatory elements of them, particularly for genes in MAPK and PI3K/AKT pathways. Among these target genes, filamin C was positively regulated by ATF3 and associated with a more favorable thyroid cancer prognosis, while dual specificity phosphatase 10, fibronectin-1, tenascin C, and CREB5 were negatively regulated by ATF3 and associated with a poorer prognosis.
Conclusions:
We observed that the promoter DNA hypermethylation decreased the expression of ATF3, which in turn promoted the progression of thyroid cancer, at least partially, by directly regulating prognosis-related genes in the MAPK and PI3K/AKT pathways.
Introduction
Many patients with thyroid cancer harbor genetic variants in the MAPK and PI3K/AKT pathways, suggesting a key role of these two signaling pathways in the pathogenesis of thyroid cancer. 1,2 Epigenetic alterations have also been reported to be involved in thyroid cancer progression. 3 However, the potential role of transcription factors (TFs) in the development and progression of thyroid cancer is largely unknown.
Activating transcription factor 3 (ATF3) belongs to the ATF/cAMP-responsive element-binding protein (CREB) TF family, which shares the basic-region leucine zipper (bZIP) domain and binds to the cAMP-responsive element site in promoters. 4,5 As a TF, ATF3 has been reported to homodimerize and repress transcription 6 or form heterodimers through its bZIP region with other ATF/CERB family proteins to exert either transcriptional repressor or activator function, depending on the cellular condition and promoter context. 4,6,7 It has been characterized as an adaptive-response gene that is involved in cellular processes to adapt to extra- and/or intracellular changes and stress responses, immune regulation, and participates in human diseases, including several types of cancer. 7 –9
ATF3 may play an important role in cancer development and progression. ATF3 interacts with p53 and increases its stability by blocking its ubiquitination in the genotoxic response. 10 However, increased ATF3 expression attenuates p53-dependent senescence and enhances the tumorigenic potential of skin squamous cancer. 11 Moreover, ATF3 has been reported to positively regulate several marker genes of epithelial–mesenchymal transition and mediate transforming growth factor-beta induced metastasis of breast cancer cells. 12,13 It was also reported to suppress RAS-stimulated tumorigenesis and act as a tumor suppressor by limiting the proliferation and invasion of lung and prostate cancer cells. 14 –16 We comprehensively investigated the regulation, function, and downstream targets of ATF3 in thyroid cancer.
Materials and Methods
Clinical samples
Eighty papillary thyroid cancer (PTC) tissue samples were obtained from a convenience sample of patients who underwent thyroid surgery between 2017 and 2019 in the First Affiliated Hospital of Sun Yat-sen University. Written informed consent was obtained from all patients for the use of their tissue samples for this study. The study was approved by the institutional ethics committee (IEC) of the First Affiliated Hospital of Sun Yat-sen University ([2021]109). The patients were all diagnosed with PTC based on postoperative histopathology, according to the criteria defined by World Health Organization. 17
The study was carried out in accordance with the ethical principles of the Declaration of Helsinki. Criteria for patient inclusion and exclusion are described in the Supplementary Data and a patient recruitment flow diagram is shown in Supplementary Figure S1. The clinicopathological characteristics of the enrolled patients are reported in Supplementary Table S1. In addition, 510 PTC samples with a median follow-up time of 31 months from The Cancer Genome Atlas (TCGA) thyroid cancer (THCA) database were included to investigate the association of ATF3 and its target genes with thyroid cancer clinical outcomes.
Among these samples, 53 progressed during the follow-up period. Specifically, 27 samples had locoregional recurrence, 15 had distant metastasis, 4 had new primary tumor, 4 died without new tumor event, 2 had biochemical evidence of progression, and 1 had new tumor event.
Cell lines and functional analyses
Thyroid cancer cell lines BCPAP, TPC-1, KTC-1, ACT-1, C643, CAL62, Hth7, KHM-5M, TTA-1, and thyroid follicular epithelial cell line Nthy-ori 3–1 were purchased from the Cell Bank of Type Culture Collection of Chinese Academy of Science (Shanghai, China). These cell lines were used for detecting the methylation and expression of ATF3, gene overexpression or knockdown, and subjected to a series of functional assays, including cell proliferation, colony formation, cell migration and invasion, and luciferase reporter assay. The details of cell culture, DNA methylation and expression analyses, and cell-based functional assays are available in Supplementary Data and the primers used in the study are listed in Supplementary Table S2.
DNA pull-down and mass spectrometry
The methylated and nonmethylated biotinylated probes, nonbiotin-labeled DNA products (5 μg), and nuclear protein extracts (500 μg) were mixed and incubated at 4°C overnight. And 50 μL of Dynabeads™ MyOne™ Streptavidin (No. 65601; ThermoFisher) was added into the mixture and incubated at 4°C for 1 hour to form DNA–protein–beads complexes. Then the pulled-down proteins were identified using liquid chromatography-tandem mass spectrometry. The methods in detail are available in Supplementary Data.
High-throughput sequencing
For RNA sequencing (RNA-seq), total RNA isolated from cells was reversely transcribed into the complementary DNA library and sequenced by Illumina NovaSeq 6000 (Illumina, USA) with paired-end 150 bp. The chromatin immunoprecipitation (ChIP)-seq assay was performed using the SimpleChIP Plus Enzymatic Chromatin IP Kit (No. 9005; CST) according to the manufacturer's protocol.
The resultant DNA by anti-ATF3 antibody, along with the corresponding 2% input DNA sample, was used to construct the ChIP-seq libraries, and the library products were sequenced by Illumina NovaSeq 6000. The CUT&Tag assay was conducted using the NovoNGS® CUT&Tag 3.0 High-Sensitivity Kit (No. N259-YH01; Novoprotein Scientific Inc.). The detailed methods of RNA-seq, ChIP-seq, and CUT&Tag assay are described in Supplementary Data.
Xenograft tumorigenicity assay
The animal experiments were approved by the IEC of the First Affiliated Hospital of Sun Yat-sen University ([2021]746). The four-week-old female nude mice were purchased from the animal center of Sun Yat-sen University and housed in specific pathogen-free facilities. The mice were randomly divided into two groups (n = 6 per group) according to random numbers generated by the RAND function of the Microsoft Excel software. The KHM-5M cells (5 × 106 cells suspended in 100 μL phosphate-buffered saline) with ATF3 overexpression or the empty vector were subcutaneously injected into the flanks of each mouse.
None of the mice was excluded from analyses. Tumor size in each mouse was measured once a day using a caliper where the tumor was visible. The tumor volume was calculated using the formula (width 2 × length) × 0.5. Then, the mice were euthanized by cervical dislocation after anesthesia at two weeks after injection. Tumors were surgically extirpated, photographed, and weighed. The investigators performing randomization and tumor cell injection were aware of the group allocations. The investigators performing the data collection and analyses were different researchers in a blinded manner.
Statistical analyses
The Shapiro–Wilk normality test and Levene's test for homogeneity of variance were used to assess the model assumptions. Two-tailed Student's t-test was applied to determine the significance of the difference between two groups. The associations between ATF3 expression and clinicopathological characteristics were analyzed by using chi-squared tests. Survival analyses according to ATF3 status were conducted using a Kaplan–Meier curve and the Cox proportional hazard regression analysis (R package survminer version 0.4.9).
The following variables were adjusted in the cox regression analysis model: age, sex, multilocality, extrathyroidal extension, lymph metastases, and history of neoadjuvant treatment. And other analyses were performed using SPSS (version 26.0; IBM SPSS) or GraphPad Prism (version 7). Two-sided p-values <0.05 were considered statistically significant.
Results
DNA hypermethylation-induced decreased expression of ATF3 in thyroid cancer
To explore the role of ATF3 in thyroid cancer, we first investigated the relative expression of ATF3 by analyzing TCGA database. As shown in Figure 1A, ATF3 was found to be significantly downregulated in 510 PTC samples when compared with the 58 normal cases (p < 0.001). A similar pattern was observed in 58 paired PTC and adjacent normal samples (p < 0.001; Fig. 1B). Then, we detected the expression of ATF3 in paired PTC and adjacent normal tissues from 80 patients in our hospital and found that 54 cases (67.5%) showed a significant decrease (Fig. 1C).

DNA hypermethylation induced reduction in expression of ATF3 in thyroid cancer. (
Moreover, the level of ATF3 was significantly lower in patients with anaplastic thyroid cancers than PTC (Supplementary Fig. S2A). We next assessed the association of ATF3 expression with clinical outcomes of thyroid cancer, and found that patients with high ATF3 expression had a longer progression-free survival (PFS) with a hazard ratio (HR) of 0.53 ([95% confidence interval {CI} 0.29–0.98], p = 0.034; Fig. 1D), and this remained significant after adjusting multiple clinical factors (HR = 0.50 [CI 0.26–0.98], p = 0.043; Supplementary Fig. S2B). Taken together, ATF3 was significantly decreased in thyroid cancer and associated with a more favorable survival prognosis.
As promoter hypermethylation is one of the major mechanisms causing gene transcriptional repression, we next evaluated whether ATF3 was regulated by DNA methylation. After searching potential CpG islands (CGIs) in the promoter region of ATF3 by MethPrimer, we found that there were five predicted CGIs (Fig. 1E). Then, according to the ATF3 transcriptional levels in thyroid cancer cell lines (Fig. 1F), TPC-1 and C643 cells with relatively low ATF3 expression as well as BCPAP, Hth7, and TTA-1 with relatively high ATF3 expression, together with Nthy-ori 3–1 cell, were randomly selected for methylation-specific polymerase chain reaction (PCR) assay to determine methylation status.
The results revealed that CGI-1 and CGI-2 of ATF3 were hypermethylated in TPC-1 and C643 cells, while the other three CGIs were unmethylated in all the cells we tested (Fig. 1G). Then, we assessed the detailed methylation status of the individual CpG sites within ATF3 CGI-1 and CGI-2 regions by bisulfite sequencing PCR. Consistently, hypermethylation was observed in TPC-1 and C643 cells (Supplementary Fig. S3A, B).
To test whether promoter DNA methylation was involved in the epigenetic silencing of ATF3, TPC-1 and C643 cells were treated with DNA demethylating agent decitabine (DAC), and it was shown that the methylation levels of CGI-1 and CGI-2 in the ATF3 promoter were significantly decreased (Fig. 1H; Supplementary Fig. S3C, D), and the expression of ATF3 was correspondingly increased after DAC treatment (Fig. 1I). Consequently, promoter hypermethylation mediated the epigenetic silencing of ATF3 in thyroid cancer.
DNA hypermethylation prevented binding of SP1 and MYC-MAX to ATF3 promoter
To further explore the mechanism by which promoter hypermethylation represses the transcription of ATF3, DNA pull-down assays were performed to screen the potential binding TFs in CGI-1 and CGI-2. We initially identified 104 and 111 candidate proteins binding specifically to the unmethylated CGI-1 and CGI-2, respectively (Supplementary Table S3). Next, we performed an in silico analysis to predict the potential TFs binding to the two CGIs and then combined them with the candidates from DNA pull-down assays. As a result, we identified 7 and 12 TFs that might be capable of binding to ATF3 CGI-1 and CGI-2, respectively.
Among these candidates, we selected SP1, MYC, MAX, USF1, and USF2 for validation (Fig. 2A). Knockdown of SP1, MYC, and MAX significantly decreased the expression of ATF3 at the transcriptional level in two cell lines with ATF3 hypomethylation (Fig. 2B, C), whereas USF1 or USF2 knockdown had no such effect (Supplementary Fig. S4A, B). In addition, the regulatory effects of SP1, MYC, and MAX on ATF3 were validated by luciferase assays (Fig. 2D).

DNA methylation blocked the binding of SP1 and MYC-MAX to ATF3 promoter. (
As shown in Supplementary Figure S5, considering that there were several potential SP1 binding sites in ATF3 CGI-1 and several MYC-MAX binding sites in CGI-2, respectively, we next performed ChIP assay to confirm the binding of these TFs to ATF3 promoter, and found that there were significantly increased occupancies of SP1 in the CGI-1 and MYC and MAX in the CGI-2 region in the hypomethylated cell line KHM-5M (Fig. 2E). In contrast, in the cells with hypermethylation of ATF3, the occupancies of SP1, MYC, and MAX were relatively low but dramatically enriched after treatment by DAC (Fig. 2F). Collectively, ATF3 promoter hypermethylation blocked SP1 and MYC-MAX induced transcriptional activation of ATF3.
ATF3 functioned as a tumor suppressor in thyroid cancer
To explore the biological function of ATF3 in thyroid cancer, we manipulated the expression of ATF3 by gene overexpression and knockdown in several thyroid cancer cell lines (Supplementary Fig. S6A–D), and the effects on cell phenotype were investigated. The results revealed that overexpressing ATF3 significantly inhibited cell proliferation (Fig. 3A), colony formation (Fig. 3B), and migration and invasion of thyroid cancer cells (Fig. 3C–E).
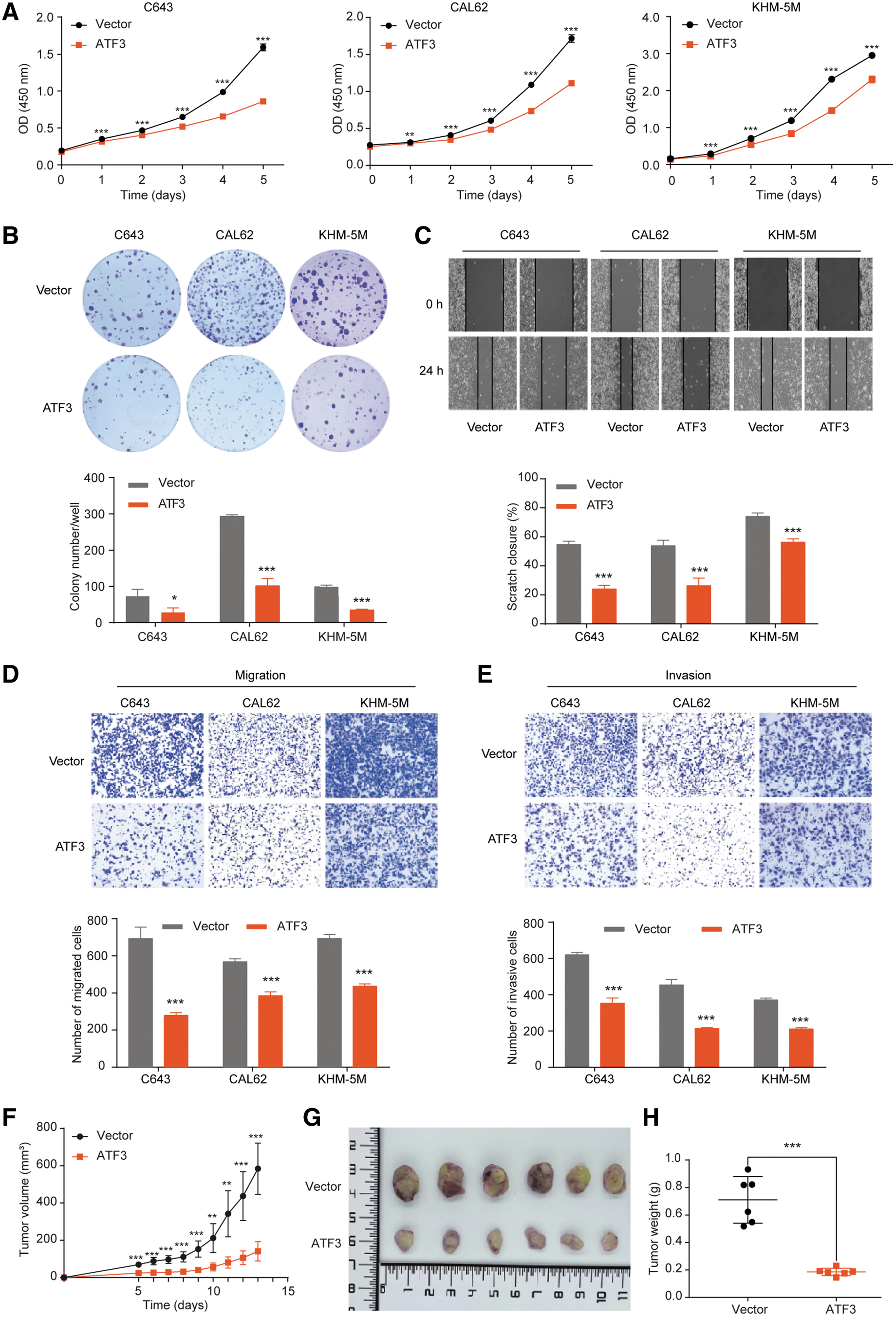
ATF3 overexpression inhibited the aggressive behavior of thyroid cancer cells. (
To address the function of ATF3 in vivo, we generated xenograft mice models using KHM-5M cells with or without ATF3 overexpression, and found that overexpressing ATF3 significantly inhibited tumor growth (Fig. 3F, G) and tumor weight (Fig. 3H) in nude mice. In contrast, inhibiting ATF3 significantly boosted the aggressive characteristics of thyroid cancer cells (Supplementary Fig. S6E–H). Collectively, these data suggested that ATF3 functions as a tumor suppressor in the progression of thyroid cancer.
ATF3 preferred to target and regulate genes in MAPK and PI3K/AKT signaling
To study the mechanism underlying the function of ATF3, ChIP-seq, CUT&Tag assay, and global transcriptome analysis (RNA-seq) were performed to explore its target genes and related signaling pathways. As shown in Figure 4A, the majority of ATF3 binding sites were near transcription start sites, suggesting that the major role of ATF3 was transcriptional regulation. The ChIP-seq identified 10,781 high-confidence binding peaks in 5091 genes, 31.75% of the peaks were located at promoters within a distance ≤1 kb (Fig. 4B).
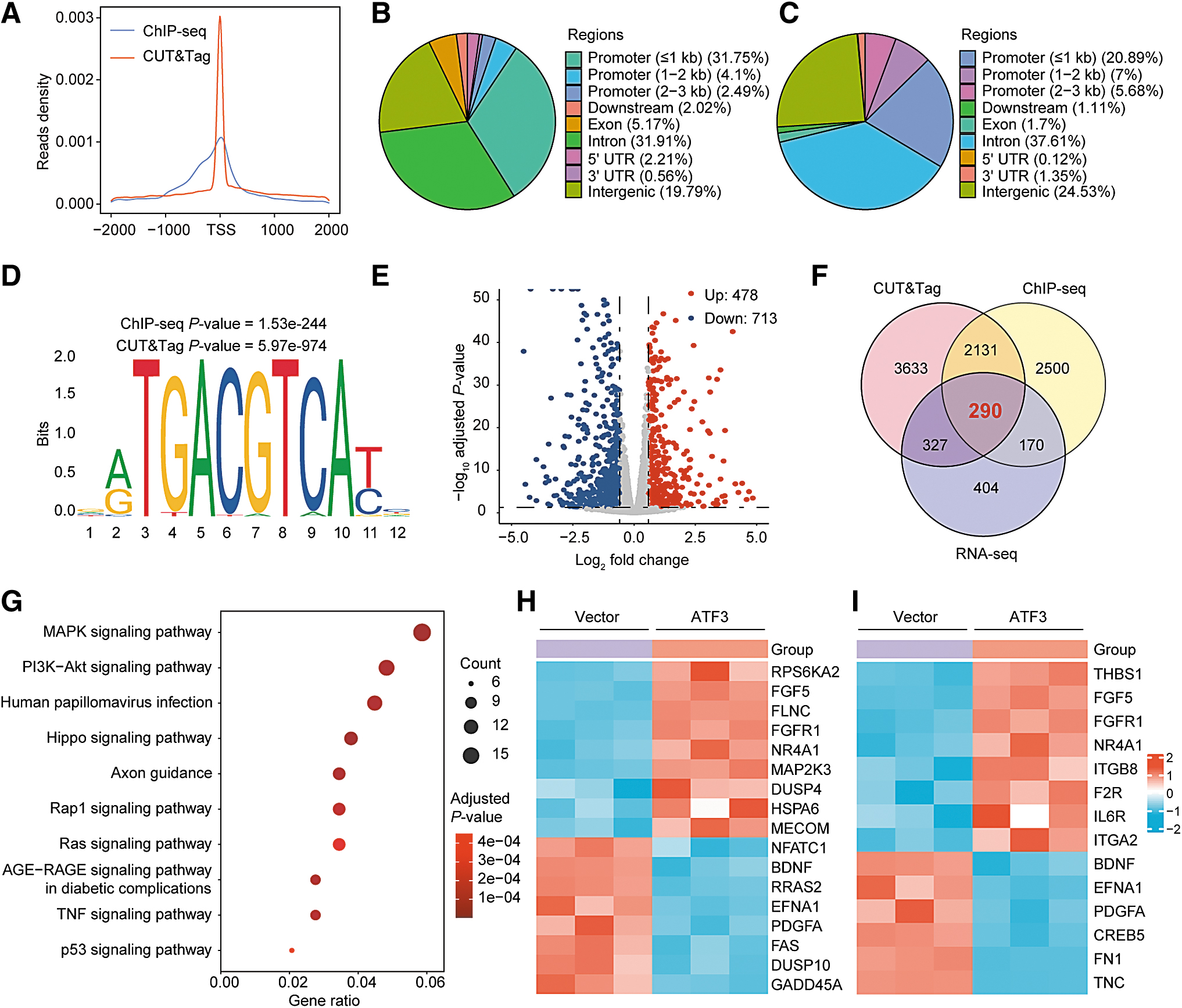
ATF3 bound to and regulated genes in MAPK and PI3K/AKT pathways. (
Among 50,159 binding peaks in 6381 genes identified by CUT&Tag, 20.89% of the peaks resided in promoters within a distance ≤1 kb (Fig. 4C). Motif analysis of these peaks revealed significant enrichment of the consensus ATF3 binding motif (Fig. 4D), and a number of de novo motifs were also identified (Supplementary Fig. S7A). Furthermore, the previously reported ATF3 binding sites in the promoter of ATF3, 18 PPP1R15A, 19 and ID1 20 were captured, and successfully verified by ChIP-quantitative PCR (qPCR) (Supplementary Fig. S7B, C).
By performing RNA-seq, we identified 478 upregulated and 713 downregulated differentially expressed genes (DEGs) in cells with ATF3 overexpression when compared with the control cells (Fig. 4E). Importantly, there were 290 DEGs overlapped with the ATF3 target genes identified by ChIP-seq and CUT&Tag assays (Fig. 4F). Gene enrichment analysis revealed that the overlap genes were enriched in several cancer-related signaling pathways (Fig. 4G), including the MAPK and PI3K/AKT pathways, which are regarded as key pathways in thyroid cancer progression. 1 As shown in Figure 4H and I, there were 17 and 14 ATF3 target genes enriched in the MAPK and PI3K/AKT pathway, respectively.
Exploration of ATF3 targets that associated with thyroid cancer prognosis
To identify the key mediator in the progress of thyroid cancer induced by ATF3, we next analyzed the expression and prognostic value of the 17 and 14 genes in the two signaling pathways. Among the 17 MAPK pathway-related genes, dual specificity phosphatase 10 (DUSP10), which was negatively regulated by ATF3, was associated with poor PFS of thyroid cancer. While the filamin C (FLNC) gene, which was positively regulated by ATF3, was associated with favorable PFS of thyroid cancer (Fig. 5A; Supplementary Fig. S8A–C).
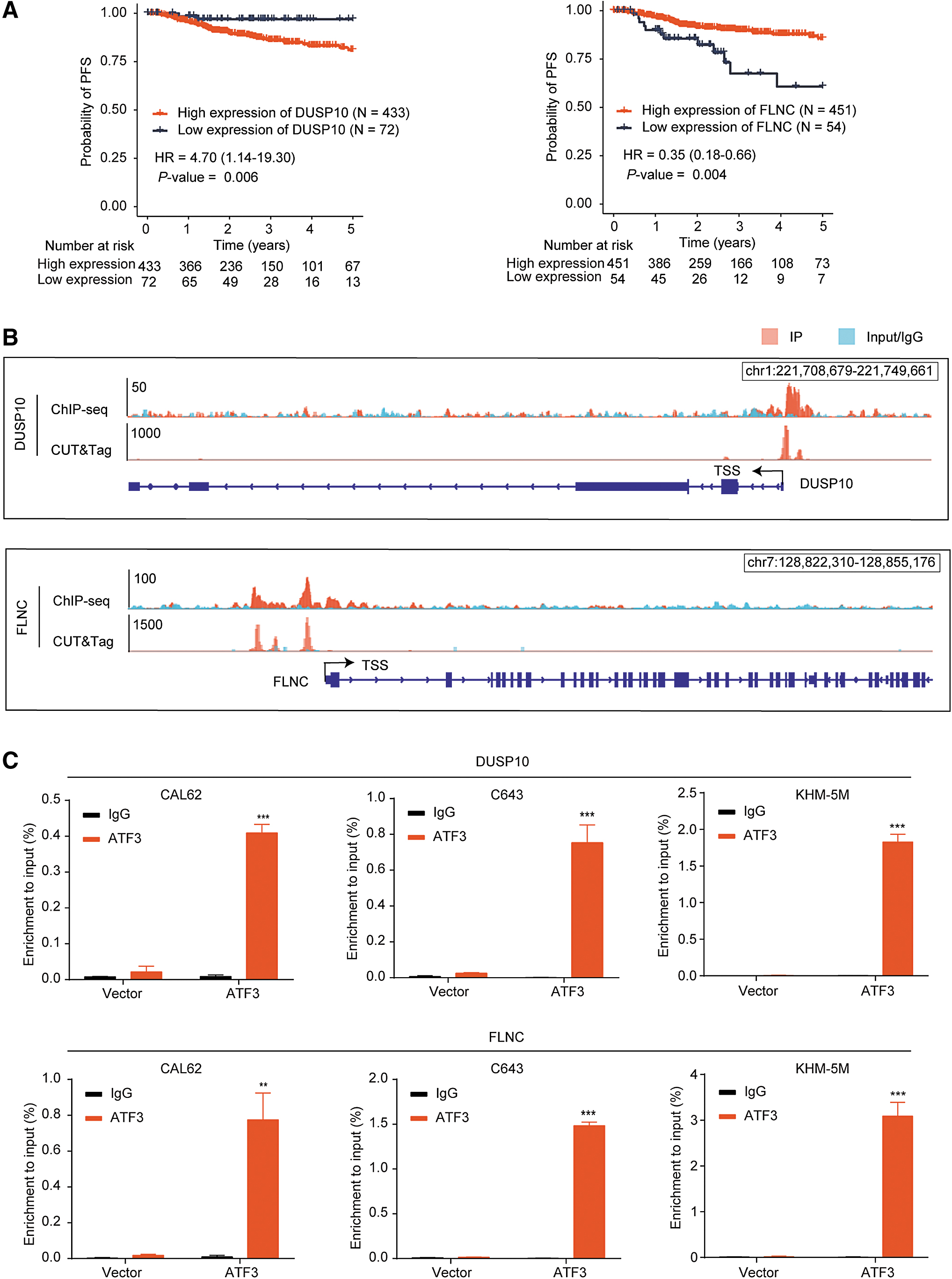
Two prognosis-related genes, DUSP10 and FLNC, in the MAPK pathway were directly regulated by ATF3. (
ChIP-seq and CUT&Tag assays showed that ATF3 was capable of binding to the promoters of DUSP10 and FLNC (Fig. 5B), which were further confirmed by independent ChIP-qPCR assays in three cell lines (Fig. 5C). Among the 14 PI3K/AKT pathway-related genes, we found that 3 genes, fibronectin-1 (FN1), tenascin C (TNC), and CREB5, were negatively regulated by ATF3 (Fig. 4I; Supplementary Fig. S8D–F) and associated with poor prognosis of thyroid cancer (Fig. 6A; Supplementary Fig. S8A). There were remarkable enrichments of ATF3 in the promoter of these three genes (Fig. 6B, C). Taken together, these data suggested that the tumor suppressing effects of ATF3 were mediated by downregulating DUSP10, FN1, TNC, and CREB5 and upregulating FLNC in thyroid cancer.
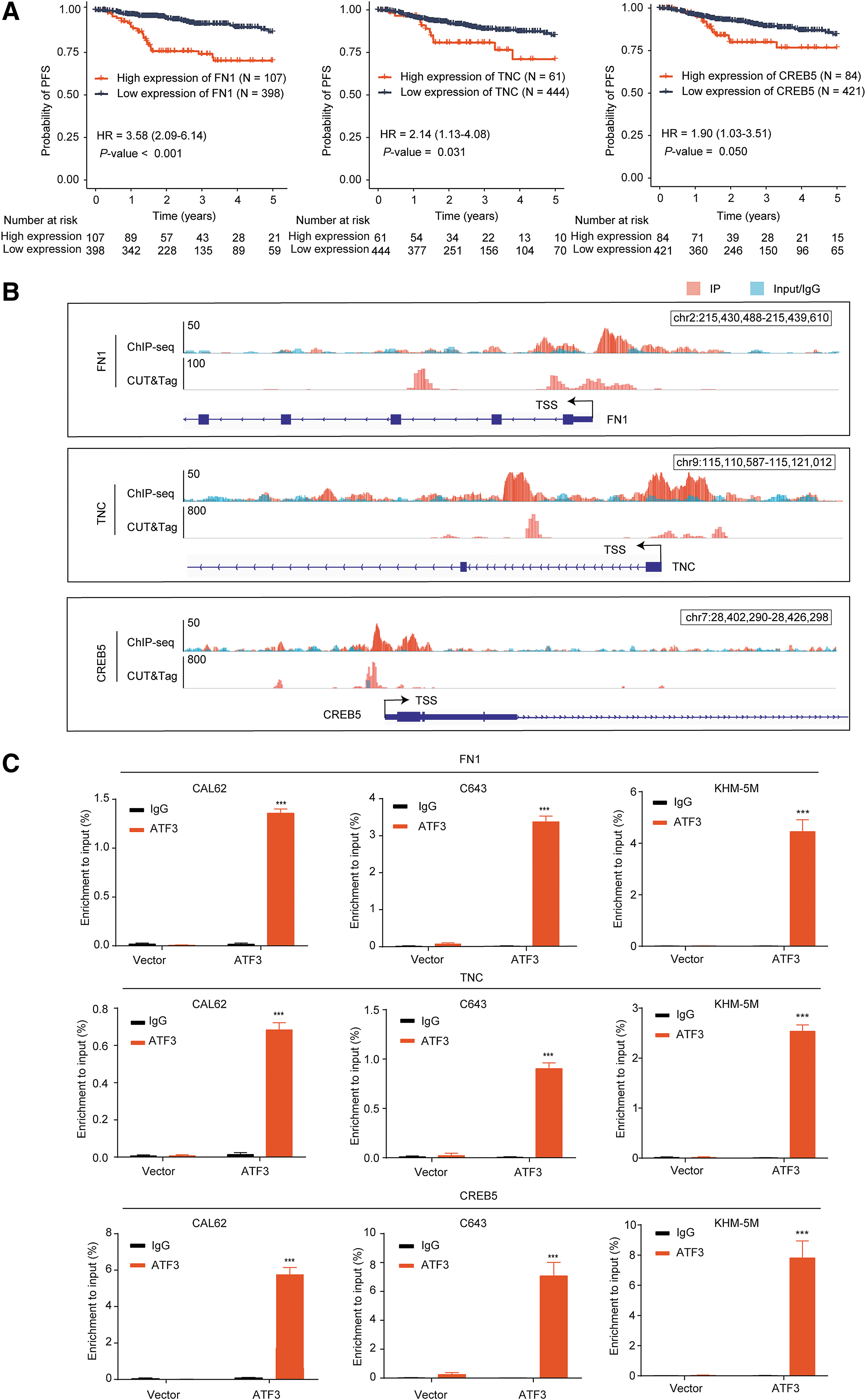
ATF3 targeted FN1, TNC, and CREB5 in the PI3K/AKT pathway. (
Discussion
As key players in the process of gene expression, TFs are commonly dysregulated in human cancers through genetic or epigenetic mechanisms. 21 In this study, we identified a member of the ATF/CREB TF family, ATF3, as a tumor suppressor and revealed that DNA hypermethylation disrupted the binding of SP1 and MYC-MAX to the promoter region of ATF3 and thus decreased its expression in thyroid cancer. Moreover, we found that ATF3 regulated the expression of a number of prognosis-related genes in the MAPK and PI3K/AKT pathways by binding to their promoters.
ATF3 was found to play contradictory roles in different types of human cancer. In our study, ATF3 was significantly downregulated in thyroid cancer and patients with low ATF3 expression had shorter PFS. Moreover, ATF3 could inhibit the growth, migration, and invasion of thyroid cancer cells. These findings are consistent with previous reports in lung and prostate cancer cells, 14 –16 suggesting a tumor suppressor role of ATF3 in these cancer types.
Although a recent study indicated that DNA methylation might involve in ATF3 regulation in breast cancer, 22 the mechanism of how methylation regulates ATF3 was unknown. Here, we determined that DNA methylation of 2 CGIs in ATF3 promoter played a key role in regulating its expression in thyroid cancer, since DAC treatment significantly decreased the methylation level of the 2 CGIs and transcriptionally restored the level of ATF3. Since CpG methylation was found to directly repress transcription by preventing the binding of TFs to their recognition motifs, 23 we conducted a DNA pull-down assay and discovered that SP1 and MYC-MAX bound to the unmethylated CGI-1 and CGI-2 of ATF3, respectively.
SP1 is a ubiquitous transcriptional activator and involves in a variety of biological processes. 24 Previous studies reported that the transactivation ability of SP1 was suppressed by the DNA methylation of its target genes, such as KIBRA and ZNF132. 25,26 MYC generally exerted physiological functions by heterodimerizing with MAX through the HLHZip domain. The heterodimer complex of MYC-MAX bound to E-box motif CACGTG in the corresponding target genes, thereby regulating the transcription of a large number of specific genes. 27,28
In fact, the potential binding sites of MYC-MAX in the ATF3 promoter have been predicted, 29 and MYC was reported to transcriptionally activate ATF3 in preconditioning peripheral nerve injury 30 and serum-induced cell proliferation. 31 We found that the binding of MYC-MAX to ATF3 promoter was abolished by hypermethylation of the promoter region, which was in accordance with the concept that DNA methylation of CpG site within the E-box may dramatically reduce MYC-MAX binding affinity to its target genes. 32,33
Furthermore, RNA-seq, ChIP-seq, and CUT&Tag assays were combined to demonstrate ATF3 direct targets. Interestingly, the enrichment analysis showed that the direct targets of ATF3 were enriched in the MAPK and PI3K/AKT pathways, both of which are classic oncogenic pathways in thyroid cancer tumorigenesis and progression. Importantly, among multiple targets of ATF3 in the 2 signaling pathways, we identified 5 genes associated with the prognosis of thyroid cancer, including DUSP10, FLNC, FN1, TNC, and CREB5.
FLNC is a dimeric actin-binding protein that can regulate remodeling of the actin cytoskeleton. 34 It was reported that FLNC can act as a scaffold to interact with stress signaling kinases MKK4 and MKK7. 35 In this study, we found that ATF3 directly targeted and upregulated the expression of FLNC, and high expression of FLNC was associated with a more favorable PFS of thyroid cancer. These data suggested the antitumor role of FLNC in thyroid cancer, which was consistent with previous findings in gastric cancer. 36,37
In contrast to FLNC, four target genes (DUSP10, FN1, TNC, and CREB5) were suppressed by ATF3 and high expression of each of these genes was associated with poorer PFS of thyroid cancer, suggesting a potential oncogenic role in thyroid cancer. DUSP10, as one of the MAPK phosphatase members, was involved in cell proliferation, differentiation, and migration in cancers. 38 The oncogenic role of DUSP10 has been reported in esophageal squamous cell carcinoma and p53 wild-type breast cancer. 39,40 A previous study showed that CREB5 significantly activated PI3K/AKT pathway in hepatocellular carcinoma and microRNA-206 inhibited tumor progression by suppressing the PI3K/AKT pathway through CREB5. 41
FN1 was also reported to be a positive regulator of PI3K/AKT pathway in many diseases. 42 –44 TNC was also reported to activate the PI3K/AKT pathway through interacting with integrins. 45 –47 Jang and Chung 48 reported that TNC-induced AKT activation promoted cell survival and was significantly repressed by the PI3K inhibitor LY294002, indicating the PI3K-dependent of TNC-mediated AKT activation. Luo et al. 49 reported that ATF3 inhibited TNC-induced macrophage foam cell formation by repressing TLR-4. However, ATF3 was reported to promote macrophage migration and reverse macrophage polarization toward M2 phenotype by upregulating TNC. 50 These data suggest that the function of ATF3 is mediated, at least partially, by activating FLNC and suppressing DUSP10, FN1, TNC, and CREB5.
This study is subject to several limitations. First, there might be selection bias in the patient samples included from our institution. Second, the sample size was relatively small and the clinical data from the patients were retrospectively collected. Our findings need to be validated by further studies with large cohorts. Despite the above limitations, to the best of our knowledge, this is the first study to systematically explore the regulation and function of ATF3 in thyroid cancer.
In conclusion, we demonstrated that DNA hypermethylation in the CGIs of ATF3 promoter suppressed the expression of ATF3 by preventing the binding of SP1 and MYC-MAX to its promoter. The resulting decreased ATF3 promoted thyroid cancer progression, at least partially, by directly regulating prognostic genes in the MAPK and PI3K/AKT pathways. These findings may inform the pursuit of a novel therapeutic strategy for thyroid cancer.
Footnotes
Acknowledgments
The authors thank Shuai He and Kai Yu at Sun Yat-sen University Cancer Center for their excellent technical assistance.
Authors' Contributions
H.X. and R.L. designed the research. X.X., M.C., Y.C., J.X., K.J., Y.S., and L.Z. performed experiments. W.L., Y.L., and S.Y. collected tissue samples. X.X. and M.C. analyzed the data. X.X., R.L., and H.X. drafted the article. H.X. and R.L. had the primary responsibility for the whole content, and all authors read and approved the final version of the article.
Data Availability
The raw sequencing data of RNA-seq, ChIP-seq, and CUT&Tag in this study are available in Sequence Read Archive with an accession number PRJNA983555.
Author Disclosure Statement
The authors declare that they have no competing financial interests.
Funding Information
The study was supported by grants from the National Natural Science Foundation of China (Nos. 82072952 and 82271776), and the “100 Top Talents Program” of Sun Yat-sen University (No. Y61227).
Supplementary Material
Supplementary Data
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3