
Abstract
Background:
The limited availability of targeted therapies in thyroid cancer (TC) has challenged conventional treatment algorithms and has established urgency for the identification of targetable genomic abnormalities. In addition to widely adopted tissue-based next-generation sequencing (NGS), plasma-based circulating tumor DNA (ctDNA) NGS is rapidly emerging as a genomic biomarker detection method and is steadily gaining utility across solid tumors. To date, plasma-based genomic alterations in TC have not been determined. Herein, we profile potential actionable mutations detected through ctDNA in patients with TC subtypes.
Methods:
A retrospective data analysis of the Guardant Health, Inc. database was performed using the commercially available Guardant360® plasma-NGS test on TC samples from adult patients collected between 2016 and 2021. The landscape of genomic alterations and blood tumor mutation burden (bTMB) were analyzed in patients with different types of TC: anaplastic TC (ATC), papillary TC (PTC), follicular TC (FTC), oncolytic carcinoma of the thyroid (OCA), poorly differentiated TC (PDTC), medullary TC (MTC), and TC not otherwise specified (TC NOS).
Results:
Of the 1094 patients included most of the patients n = 876 had TC NOS, and 20% had a specific diagnosis (92 ATC, 62 PTC, 14 FTC, 16 OCA, 2 PDTC, and 32 MTC patients). The median age was 65 (range 10–98) and 47.3% were male. 78.3% of patients had one or more genomic alteration detected by ctDNA NGS. TP53 (46.9%) was the most common mutation detected among all TC. BRAFV600E was detected in 27.2% of ATC, 35.7% of PTC, and in none of FTC. RAS was detected in 18.5% of ATC, 11.9% of PTC, and 62.5% of FTC. RET, ALK, and NTRK fusions were seen in 1.1%, 0.5%, and 0.2% of all TC, respectively. RET mutations were detected in 66.7% of MTC. bTMB analysis was performed on 159 patients. The mean bTMB was higher in ATC compared with other types of TC (p = 0.0011, 0.0557, and <0.0001, respectively).
Conclusions:
Plasma-based comprehensive NGS is a promising NGS method in TC; however, future validation of the clinical utility by analysis of paired tumor and plasma samples is needed.
Background
Thyroid cancer (TC) represents a broad spectrum of disease that varies in cell of origin (i.e., follicular cells or C-cells), histology, and clinical outcomes. The tumors with the best prognosis include well-differentiated low-risk intrathyroidal papillary TC (PTC) and minimally invasive follicular TC (FTC). Anaplastic TC (ATC) has the worse prognosis. Medullary TC (MTC) is a distinct type of TC arising from the neuroendocrine cells (c-cells).
For all subtypes, patients with progressive metastatic TC have significantly worse prognoses and overall survival (OS). 1 –3 The prognosis and treatment of TC varies significantly based on the cancer type, stage, location, and burden of recurrent or persistent disease, rate of progression, symptoms, and other factors. The identification of specific driver alterations in ATC has enabled treatment with targeted therapies and resulted in improvement of OS. 4
Historically, multikinase inhibitors have been used as the first-line treatment where systemic therapy was indicated. 5 However, recent approval of highly selective targeted agents has challenged the conventional treatment algorithms and has established urgency for the identification of novel targetable genomic abnormalities. 6,7 These targeted agents include the combination of dabrafenib and trametinib—approved for BRAFV600E mutated ATC and solid tumors without satisfactory alternative treatment options, including differentiated TC (DTC); entrectinib and larotrectinib—approved for any cancer harboring an NTRK gene fusion, and selpercatinib and pralsetinib—approved for TC with RET rearrangements in DTC or RET mutations in MTC. 7 –10
The availability of highly effective treatments and powerful next-generation sequencing (NGS) has resulted in practice changes reflected in the National Comprehensive Cancer Network (NCCN) guidelines on TC and the American Thyroid Association (ATA) guidelines on ATC. 11,12 It has also created opportunities for neoadjuvant treatment in locally advanced TCs that were previously not feasible. For example, the combination of dabrafenib and trametinib as neoadjuvant therapy for patients with BRAFV600E mutated ATC has demonstrated improved survival. 4
In addition, early identification of RET mutations to implement neoadjuvant therapies for locally advanced DTC and MTC are currently being investigated in a clinical trial (NCT04759911). Moreover, the use of NGS has driven clinical trials, including NCT05182931, which assesses the response to emerging tyrosine kinase inhibitor (TKI) redifferentiation therapies based on the presence of a driver mutation. 13 The NCCN and ATA guidelines on ATC, DTC, and MTC endorse tumor testing for somatic mutations and gene alterations. 11,12 However, the choice of the testing methodology, timing, and sequence of genomic testing are not specified and remain unclear. 11,12,14
NGS of the primary tumor or metastatic disease is commonly used for comprehensive genomic profiling in TC. Tissue-based NGS can be challenging due to insufficient tumor availability and difficulty accessing metastatic lesions. In addition, it can be difficult to retrieve archival tissue samples as metastatic disease is frequently diagnosed years after thyroidectomy and obtaining these samples from other institutions can introduce delays in sequencing and/or additional biopsy requirements. Ultimately, these barriers to molecular testing can result in suboptimal treatment decisions, potential therapy delays, and inferior outcomes.
Consequently, alternative methods are needed to identify targetable and clinically relevant genomic alterations. One method is plasma-based genomic profiling of circulating tumor DNA (ctDNA) through broad genomic panels, including clinically actionable alterations. NGS of ctDNA has been validated in patients with solid tumors and broadly adopted for patients with lung cancer but has not been extensively studied in patients with TC. 15
Previous studies have focused on describing the clinical and prognostic impact of specific mutations detected in ctDNA from patients with TC. 4,16 –18 In patients with advanced DTC, BRAFV600E detected in ctDNA is associated with tumor aggressiveness and response to therapy. 16 In addition, the presence of RET M918T in ctDNA is correlated with worse OS in patients with MTC. 17 In patients with ATC, there were high rates of up to 96% concordance between solid tissue and ctDNA NGS. 4,19
Moreover, comprehensive ctDNA NGS has demonstrated faster turnaround time as compared with tissue NGS in ATC. 18,19 These data support the clinical utility of ctDNA in TC. 20 Therefore, in this study we describe the performance of NGS of ctDNA in patients with different subtypes of TC.
Methods
Patient characteristics
The generation of de-identified data sets by Guardant Health for research purposes was approved by the Advarra Institutional Review Board (Pro00034566). All patients signed a consent for molecular testing and research use. Patient identity was maintained throughout the study in a de-identified database. We conducted retrospective review of data included in the Guardant Health database to perform a cross-sectional analysis on adult patients with TC tested between 2016 and 2021 with Guardant360® (Guardant Health, Redwood City, CA), a plasma-based NGS assay. The type of TC was reported by an ordering provider on the Guardant360 requisition form at the time the test was ordered.
Patients with TC were analyzed based on the type of cancer: ATC, PTC, FTC, oncolytic carcinoma of the thyroid (OCA), poorly differentiated TC (PDTC), and MTC. Patients who did not have a specific type of cancer reported on the requisition form were classified as TC not otherwise specified (TC NOS) (included all types of TC: ATC, PTC, FTC, OCA, PDTC, and MTC) as summarized in Figure 1. Patients with other malignancies known at the time of the first sample collected or any consecutive samples were excluded from the study. All data were de-identified.
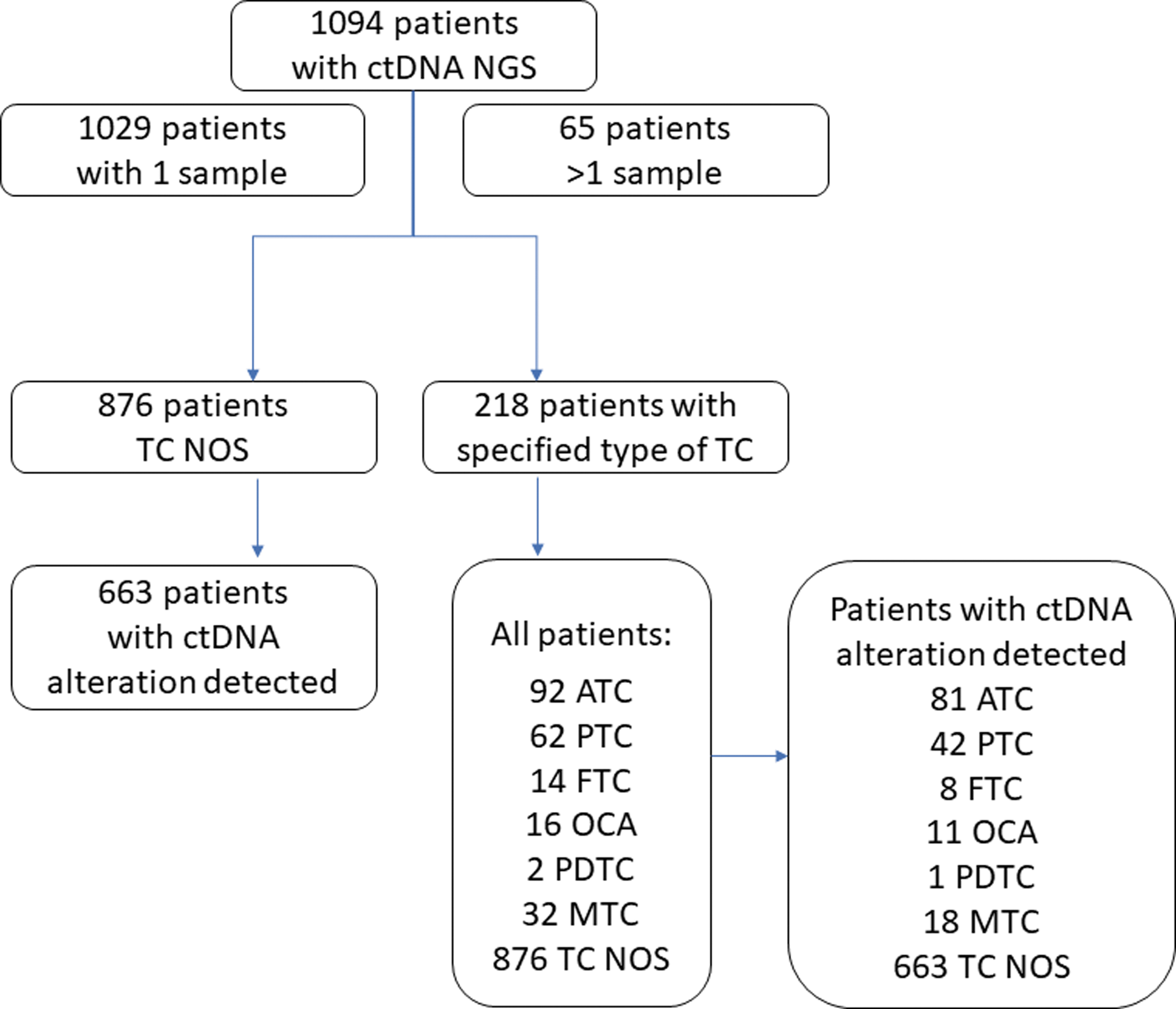
Flow diagram. ATC, anaplastic thyroid cancer; ctDNA, circulating tumor DNA; FTC, follicular thyroid cancer; MTC, medullary TC; NGS, next-generation sequencing; OCA, oncolytic carcinoma of the thyroid; PDTC, poorly differentiated TC; PTC, papillary thyroid cancer; TC, thyroid cancer; TC NOS, thyroid cancer not otherwise specified.
NGS assay characteristics
Guardant360 is an NGS assay that uses targeted high-throughput hybridization-based capture technology for the detection of clinically relevant genomic alterations from ctDNA in plasma collected through peripheral whole blood. Patients were tested with broad panels that have previously been validated for single nucleotide variants (SNV) and insertions/deletions (indel) in 70–83 genes as well as select genes for copy number alterations and fusions. 21 For this analysis, putative clonal hematopoiesis of indeterminate potential (CHIP) mutations were filtered and excluded. 22
A subset of patients tested with a more recent panel was also analyzed for blood tumor mutational burden (bTMB). bTMB was calculated using methods as previously described, analyzing somatic SNVs and indels across a 1.0 Mb genomic backbone. 22 For the bTMB algorithm, common cancer drivers and resistance alterations, as well as putative CHIP alterations were filtered from analysis. Putative germline alterations were also excluded from the analysis and separated from somatic alterations through a beta-binomial statistical model. 23
The landscape of genomic alterations with co-occurrence and mutual exclusivity of genomic alterations were analyzed. We selected specific gene alterations based on previously described genomic landscapes and genes associated with clinical relevance in TC and based on the expert opinion of the authors. 4,24 –26 The genes investigated were BRAF (BRAFV600E and others), RAS (KRAS, NRAS, and HRAS), RET, PTEN, TP53, TERT promoter, PIK3CA, NF1, ATM, RB1, STK11, and CHEK2. We also analyzed the prevalence of RET, ALK, and NTRK1/2/3 fusions.
Of note, NTRK1 fusions were reported in all panels, but NTRK2/3 fusions were only available in a recent panel available for some patients tested during and after 2020. For this landscape analysis, alterations were visualized with oncoprints using software available from cBioPortal. 27,28 For patients with serial tests, the presence of an alteration was only counted a maximum of once per patient to reduce duplicative frequency reporting in Tables 2 and 3.
Statistical analyses
Descriptive statistics were used to report the results of the study. Limited demographic, clinical, and pathological information were available for the samples. Consequently, we analyzed data according to the type of TC, age, and sex. Statistical analysis for age was conducted with an unpaired Student's t-test for each subgroup as compared with the remainder of the cohort. The statistical analysis for sex was conducted with a two-sided Fisher's exact test. A statistical analysis for bTMB scores was calculated using a two-way ANOVA. Statistical analysis was performed using GraphPad Prism version 9.0 for Windows, GraphPad Software (San Diego, CA).
Results
Characterization of genomic alterations among different types of TC
The Guardant Health database contained data from 1202 samples obtained from 1094 patients collected between 2016 and 2021. There were 1029 patients with a single sample of ctDNA NGS and 65 patients with >1 sample tested. Most patients were generally characterized as TC without details of the subtype (TC NOS n = 876 patients, 80%); however, 218 patients (20%) had a specific diagnosis and were, therefore, categorized according to the cancer type: ATC (n = 92, 8.4%), PTC (n = 62, 5.7%), FTC (n = 14, 1.3%), OCA (n = 16, 1.5%), PDTC (n = 2, 0.2%), and MTC (n = 32, 2.9%).
Demographics from patients in this cohort are reported in Table 1. The median age was 65 (range 19–98) and 47.3% of patients were male. There were no significant differences in age between any types of TC, except for patients with MTC. MTC patients were younger compared with other groups (p = 0.001). There were no significant differences in the sex within the TC types (p ≥ 0.6–11).
Patient Demographics
ATC, anaplastic thyroid cancer; FTC, follicular thyroid cancer; MTC, medullary thyroid cancer; OCA, oncolytic carcinoma of the thyroid; PDTC, poorly differentiated thyroid cancer; PTC, papillary thyroid cancer; TC NOS, thyroid cancer not otherwise specified.
A ctDNA genomic alteration (≥1) was identified in 857 patients (78.3%). A subset of genes of interest, chosen from previously published data and clinical relevance, was analyzed for frequency among each group (Table 2). Among all types of TC with one or more genomic alteration detected, the most common genomic alteration occurred in TP53 (46.9% of patients), followed by BRAF (21.3%), RAS (19.6%), and TERT promoter mutations (13.8%). RET, ALK, and NTRK fusions were seen in 1.1%, 0.5%, and 0.2%, respectively. The BRAFV600E mutation was the most common mutation identified among all patients with BRAF mutations detected (78.4%).
Genomic Alterations Detected in Thyroid Cancer
In ATC, BRAFV600E and RAS mutations were detected in 27.2% and 18.5% of patients, respectively. Among all RAS mutations in ATC, the most frequently mutated was NRAS (11.1%), followed by HRAS and KRAS (3.7% each). TP53 and TERT promoter mutations were detected in 65.4% and 17.3% of patients with ATC, respectively. RET fusions were detected in 1.2% of the ATC cohort.
Patients with PTC had BRAFV600E and RAS mutations detected in 35.7% and 11.9%, respectively. No BRAFV600E mutations were detected in patients with FTC. RAS mutations were detected in 62.5% of FTC patients. Among RAS mutations in FTC patients, 37.5% were NRAS and 25% were HRAS. No KRAS mutations were seen in FTC. TERT promoter mutation was most prevalent in patients with FTC (37.5%) compared with other types of TC. A trend of lower rates of TP53 mutations were seen in patients with PTC and FTC compared with ATC (42.9% and 50% compared with 65.4%). TERT promoter mutations were seen in this group at 16.7% and 37.5% compared with 17.3% in ATC. There was only one evaluable PDTC patient. This patient had TP53 and PTEN alterations.
In patients with MTC, RET mutations were most frequent (66.7%), followed by TP53 mutations (16.7%). KRAS and ATM mutations and ALK fusions were detected in 5.6% of patients with MTC. BRAFV600E mutations were not detected in this group. Among RET mutations detected in patients with MTC, the most common was M918T in 27.8% (Table 3). RET C634R, C634L, C634S, and V804M mutations were each detected in 5.6% of MTC patients. Table 3 reflects specific RET mutations detected by ctDNA NGS among different types of TC.
RET Mutations Detected by Circulating Tumor DNA Next-Generation Sequencing in Different Types of Cancer
ctDNA, circulating tumor DNA; NGS, next-generation sequencing.
Evaluation of co-occurring and mutually exclusive genomic alterations among different types of TC
The TC groups were evaluated for patterns of co-occurrence and mutual exclusivity among driver mutations (i.e., BRAFV600E , RAS, and RET alterations). ATC patients demonstrated mutually exclusive driver mutations for BRAFV600E and RAS (p = 0.008). In the TC-NOS group, BRAFV600E demonstrated mutual exclusivity with KRAS, NRAS, and HRAS (p ≤ 0.037). PTC demonstrated co-occurring genomic alterations for BRAFV600E and TERT promoter (p < 0.001). Similarly, BRAFV600E and TERT promoter co-occurring alterations were significantly associated with TC-NOS (p < 0.001).
Oncoprints for the distinct cancer groups were created to show the landscape of co-occurring alterations (Figs. 2 –5). Sample numbers in the oncoprints analysis differ from Table 1, as samples with nondetectable alteration values, synonymous mutations, and putative CHIP alterations were removed from this analysis. Only unique sample IDs were included in the oncoprints.

Oncoprints for anaplastic TC show landscape of co-occurring alterations. Oncoprint analysis of ATC samples. A total of 84 unique ATC samples from 81 patients were analyzed using cBioPortal for co-occurring alterations. Commonly occurring alterations in ATC samples are presented here.

Oncoprints for papillary, follicular, OCA, and PDTCs show landscape of co-occurring alterations. Oncoprint analysis of PTC, FTC, OCA, and PDTC samples. A total of 64 unique overall samples from 62 patients were analyzed using cBioPortal for co-occurring alterations. Commonly occurring alterations in PTC, FTC, OCA, and PDTC samples are presented here.
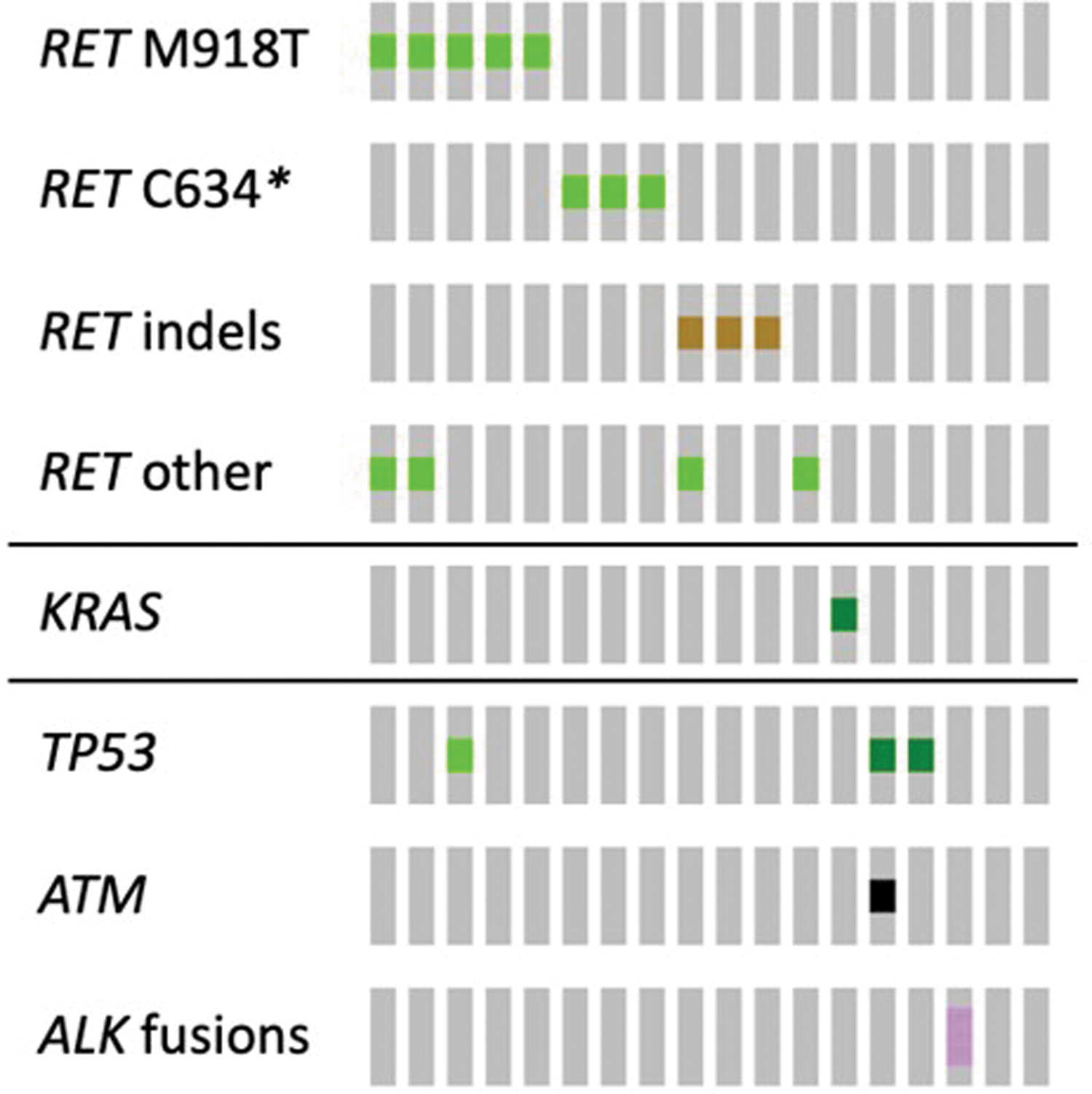
Oncoprints for MTC show landscape of co-occurring alterations. Oncoprint analysis of MTC samples. A total of 18 unique MTC samples from 18 patients were analyzed using cBioPortal for co-occurring alterations. Commonly occurring alterations in MTC samples are presented here.
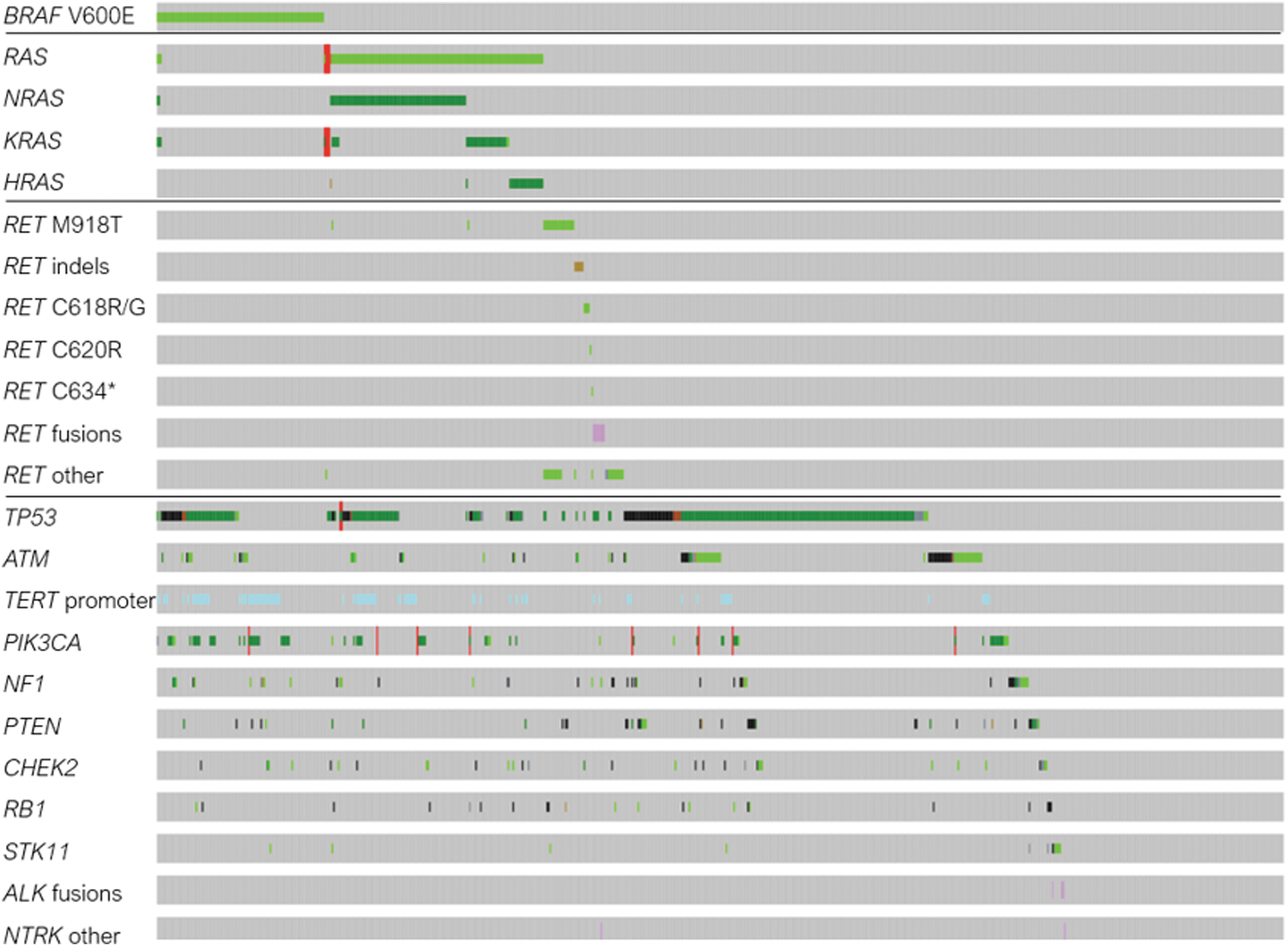
Oncoprints for nonspecified TC show landscape of co-occurring alterations. Oncoprint analysis of TC NOS samples. A total of 729 unique TC NOS samples from 663 patients were analyzed using cBioPortal for co-occurring alterations. Commonly occurring alterations are presented here.
Assessment of tumor mutation burden among different types of TC
TMB has been integrated into practice as a biomarker for response to immunotherapy across solid tumors. Thus, we sought to analyze bTMB in our cohort. bTMB analysis was performed on 164 samples from 159 patients tested with the panel measuring bTMB. Mean bTMB in ATC was 26.12 mut/Mb—this value was higher than other types of TC. Grouped together PTC, FTC, OCA, and PDTC demonstrated bTMB at 7.30 mut/Mb, MTC at 2.30 mut/Mb, and unspecified TC 8.25 at mut/Mb (p = 0.0011, 0.0557, and <0.0001, respectively; Fig. 6). Mean bTMB for each group was 7.66, 6.57, 1.91, and 6.82 mut/Mb, respectively.
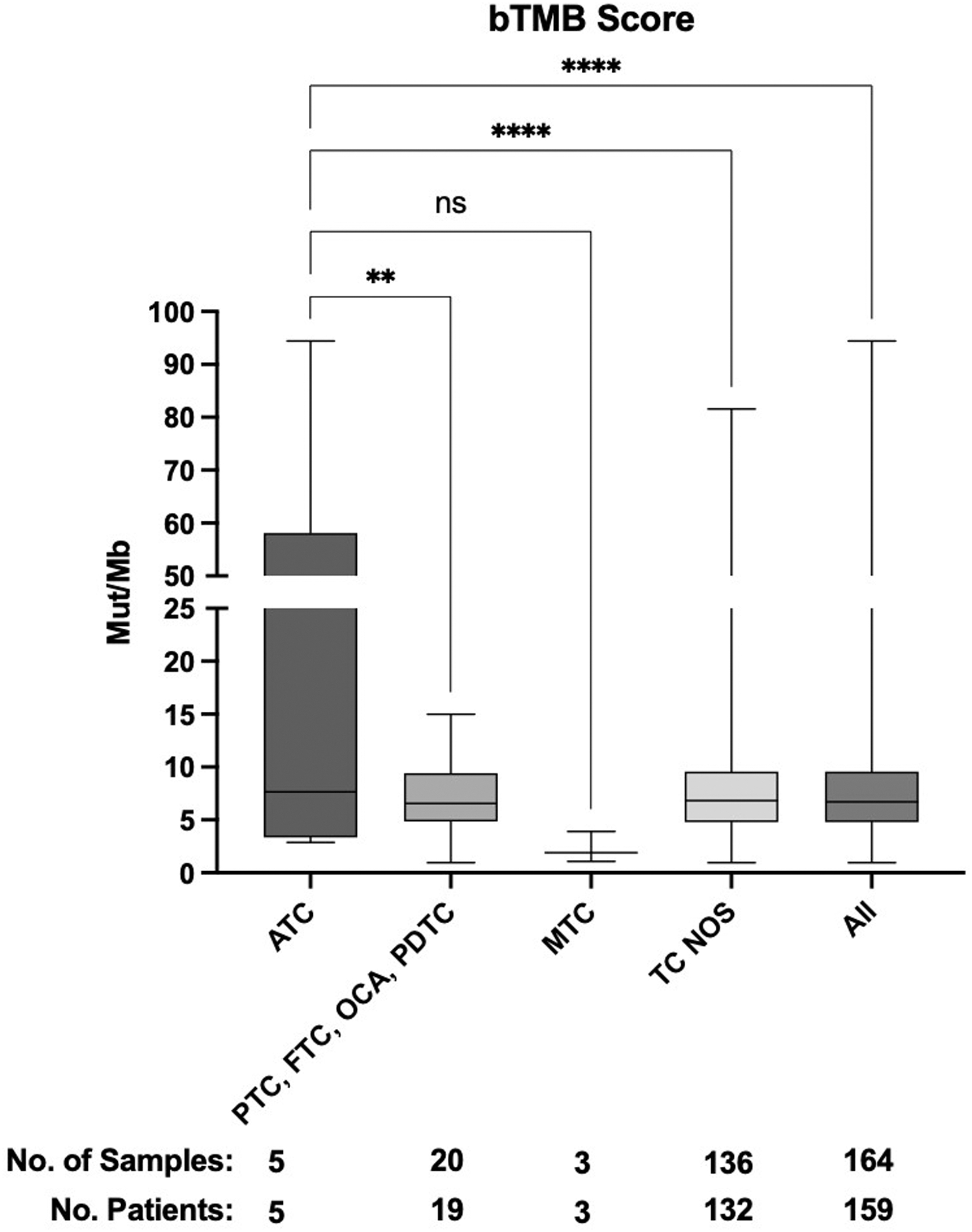
Blood tumor mutation burden among different types of TC. bTMB scores from 159 patients calculated using a two-way ANOVA. Median bTMB for each group was 7.660, 6.570, 1.910, and 6.820; **p ≤ 0.01 and ****p ≤ 0.0001. bTMB, blood tumor mutational burden; mut/Mb, mutation per megabase; ns, not significant.
Discussion
In this study, we characterized TC genomic landscape by ctDNA NGS. To date, this study describes the largest cohort of patients (n = 1094) with various TC types analyzed through ctDNA NGS. ctDNA NGS detected at least one alteration in 78.3% of patients. Previously, reported genomic alterations were identified with tissue NGS in 96.5% of DTC, in over 88.5% of ATC and 89.1% in MTC. 4,24,29 TP53 alterations were the most prevalent mutations among all TC cases in our cohort.
ATC, PTC, and FTC had higher rates of TP53 mutation detected compared with the MTC patients (65.4%, 42.9%, and 50% vs. 16.7%, respectively). Consistent with the literature, TP53 mutation was the most common mutation detected in 54% of patients with ATC by tissue NGS. 4 Among BRAF mutations, BRAFV600E was the most common mutation detected (78.4%).
The rates of detected BRAFV600E by ctDNA were lower in our study compared with historic cohorts. BRAFV600E mutation was detected in 35.7% of DTC and 27.2% ATC patients by ctDNA, compared with the 74% and 41%, respectively, by tissue NGS. 25 Lower detection rates of specific genomic alterations through ctDNA may potentially be explained by specific characteristics of TC, such as tumor burden and tumor shed. Interestingly, selected mutations detected by ctDNA were at comparable rates with tissue NGS. RAS mutations were detected in 63% of FTC by ctDNA compared with 66% by tissue NGS. 25
Similarly, RET mutation was detected in 66.7% of MTC patients, compared with 64.5% by tissue NGS. 29 Paired tissue and ctDNA NGS as well as clinical correlations may help to clarify this discordance. The analysis of ctDNA NGS confirmed previously reported data on the mutual exclusivity of the driver mutations, BRAFV600E and RAS, in PTC, FTC, and ATC. None of the detected BRAF mutations in MTC nor OCA patients were BRAFV600E mutations. There were no BRAF mutations detected in patients with FTC. All BRAF mutations detected in PTC were BRAFV600E.
RET mutation detection by ctDNA NGS in patients with MTC was very similar to tissue NGS, 66.7% versus 64.5%, respectively. 29 This supports the utility of ctDNA to capture somatic RET alterations in MTC. Approximately 25% of MTC are driven by germline RET alterations. 30 Although it is clinically essential to establish germline RET alterations in MTC, the ctDNA assay that was used in this study was only validated to identify somatic variants and not germline. Nonetheless, a RET mutation with a high allele frequency might suggest a germline mutation, and establishing germline RET mutation status on ctDNA NGS warrants further investigation.
There were no sex differences between different types of TC. This is likely because this cohort was limited to advanced metastatic TC, likely with a significant subset of radioactive iodine refractory disease. 1,31
The assessment of TMB has clinical implications due to its association with the response to immunotherapy. 32 High TMB has been associated with improved response rates to anti-programmed death 1 (PD-1) or anti-programmed death 1 ligand (PD-L1) targeted therapies among different types of cancers. 32 Tissue NGS of advanced TCs demonstrated that the accumulation of key genomic abnormalities and higher TMB were associated with more aggressive clinicopathological characteristics of TC. 25 bTMB estimated on ctDNA NGS by Guardant360 was significantly higher in patients with ATC compared with other types of TC, as expected.
The cohorts in this study demonstrated higher frequencies of certain mutations, including TP53 and KRAS, than typically found in tissue. A potential explanation for this finding is that plasma-based NGS may have detected CHIP; although they were filtered and excluded with the best of our ability. 33 An additional explanation for TP53 is that patients with advanced TCs are known to have a high prevalence of TP53 mutation. 25
In addition, the assay used in this study is directed toward patients with advanced stage cancer, which tend to have increased ctDNA shed as compared with early-stage cancer. 34 The cohorts in this study also demonstrated higher frequencies of BRAF alterations. As CHIP mutations do not typically occur in BRAF gene, the substantial subset of non-BRAFV600E mutations detected by ctDNA NGS among TC patients warrants further investigation.
Our study has several major limitations, including a lack of confirmation with the tissue NGS analysis. Therefore, the sensitivity and specificity of the ctDNA and concordance rates between tissue NGS and ctDNA NGS cannot be determined in this study.
Another limitation is related to limited availability of clinicopathological data on the patients and information regarding other genomic tests performed. Only 20% of patients had TC type reported. This may be related to the universal C73 code for all types of TC used for billing based on the International Classification of Diseases (ICD) classification. We assume that the study participants had distant metastatic disease or locally advanced TC, as these are the patients who qualify for genomic testing based on guidelines. 11
There is no information on the timing of the ctDNA testing used for these patients. Consequently, we hypothesize that some samples were taken at initial diagnosis, some at the time of progression, and others to assess therapy response. Therefore, in addition to the driver genomic alterations, some mutations associated with resistance to therapy may have been detected, although there are not clinical data in this study to support this.
One of the primary mechanisms of resistance to TKIs in TC is the acquisition of secondary mutations that are downstream of the primary oncogenic driver mutations. 35 Therefore, we hypothesize that patients with PTC and TC NOS group with both BRAFV600E and RAS (KRAS and NRAS) mutations detected may have developed the latter as a mechanism of resistance to TKIs (Figs. 3 and 5). In addition, the largest patient group, “TC NOS” (n = 876), who did not have TC type described, may have included all types of TC: ATC, DTC, PDTC, and MTC.
The selection, sequencing, and timing of molecular testing in patients with TC remain uncertain. Many factors to be considered including the availability of genomic tests, expertise of the specialties/facilities performing the tests, and quality and quantity of the tissue as well as the feasibility of obtaining the result in a clinically reasonable turnaround time, the cost of testing, and insurance coverage. Therefore, it is important to educate the providers and patients on this topic.
Our data support that ctDNA NGS can complement tissue NGS in patients with TC although further studies are warranted with comprehensive analyses of both tissue and ctDNA NGS. ctDNA NGS is a minimally invasive detection tool for biomarkers that can be considered as an alternative method to tissue NGS in patients with advanced TC in whom tissue NGS cannot be performed. In addition, further investigations are required to overcome the current barriers in molecular testing and delineate the role of ctDNA NGS, which can have potential implications in establishing diagnosis, determining prognosis, and understanding resistance mechanisms in patients with TC.
Moreover, quantitative assessment of ctDNA may play a role in determining the response to therapy and the rate of progression as well as potentially serving as an early marker for recurrence, particularly in patients with ATC, in whom tumor marker (i.e., thyroglobulin) is not reliable. 12
To date, this study describes the largest cohort of patients (n = 1094) with TC who have undergone ctDNA NGS. This study reflects the real-life experience of detecting genomic alterations in patients with TC. Clinical correlations of genomic alterations detected by ctDNA and histological type, stage, location of metastasis, burden of the disease, rate of progression, and response to therapy are warranted. The performance of ctDNA NGS to detect actionable driver mutations and genomic alterations to select treatment warrants further investigation by prospective studies.
Footnotes
Authors' Contributions
Conceptualization of the study idea and design, writing—original draft (lead), review, and editing the article (lead) by V.D.T. Writing—original draft, review, and editing (lead), data analysis (lead), and statistical analysis (lead) by J.T. Data analysis, statistical analysis, created oncoprints, and writing—review and editing by J.M. Data interpretation and writing—review and editing by J.C.H.P., J.H.J., C.V., and S.A.S. Conceptualization of the study idea and design, data review, and editing by B.M. Conceptualization of the study idea and design, methodology, and writing—review and editing by L.M.D. Supervising the research project, data interpretation, and writing—review and editing by C.H.C.
Author Disclosure Statement
V.D.T., J.H.-J., J.C.H.-P., and C.V. have no relevant conflicts of interest to disclose. S.A.S. has served on advisory boards for Eisai, Exelixis, and Blueprint Medicine and consulting/speaker for Eli Lilly. Bryan McIver serves on advisory boards for Eisai, Blueprint, and Beyer, and has received speaker honoraria from Eisai, Eli Lilly, and Sonic Healthcare USA. C.H.C. has served in Exelixis Scientific advisory board. J.T., J.M., and L.M.D. are employees and stockholders of Guardant Health, Inc. The abstract was presented at ASCO in 2022.
Funding Information
No funding was received or will be received in the future for this article by non-Guardant Health, Inc. employees (V.T., J.C.H.P., J.H.J., C.V., S.A.S., B.M., and C.H.C.). Guardant Health, Inc. employees (J.T., J.M., and L.M.D.) receive salary from the company. Database was developed and maintained by Guardant Health, Inc.