
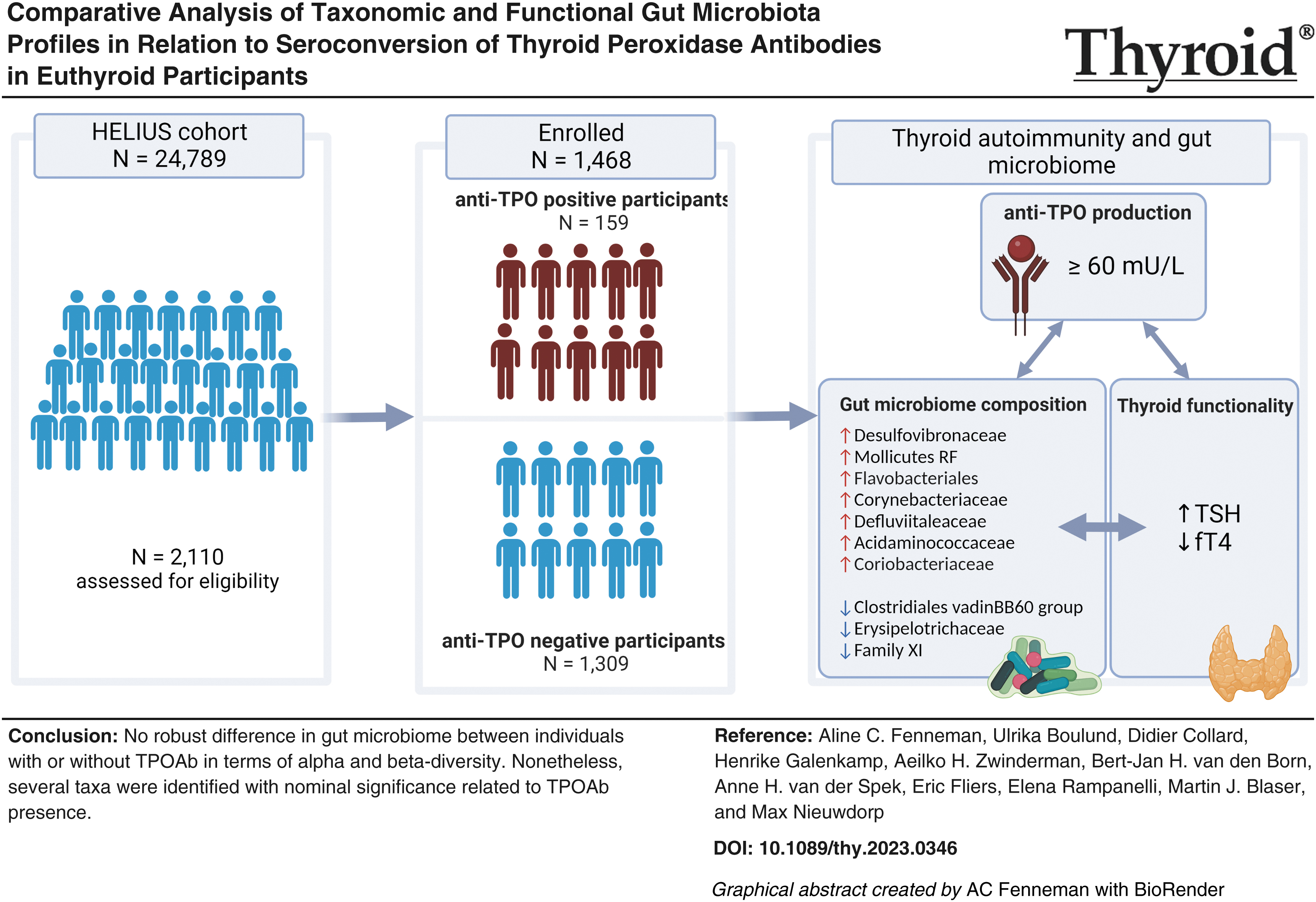
Abstract
Background:
Previous studies have reported gut microbiome alterations in Hashimoto's autoimmune thyroiditis (HT) patients. Yet, it is unknown whether an aberrant microbiome is present before clinical disease onset in participants susceptible to HT or whether it reflects the effects of the disease itself. In this study, we report for the first time a comprehensive characterization of the taxonomic and functional profiles of the gut microbiota in euthyroid seropositive and seronegative participants. Our primary goal was to determine taxonomic and functional signatures of the intestinal microbiota associated with serum thyroid peroxidase antibodies (TPOAb). A secondary aim was to determine whether different ethnicities warrant distinct reference intervals for accurate interpretation of serum thyroid biomarkers.
Methods:
In this cross-sectional study, euthyroid participants with (N = 159) and without (N = 1309) TPOAb were selected from the multiethnic (European Dutch, Moroccan, and Turkish) HEalthy Life In an Urban Setting (HELIUS) cohort. Fecal microbiota composition was profiled using 16S rRNA sequencing. Differences between the groups were analyzed based on the overall composition (alpha and beta diversity), as well as differential abundance (DA) of microbial taxa and functional pathways using multiple DA tools.
Results:
Overall composition showed a substantial overlap between the two groups (p > 0.05 for alpha-diversity; p = 0.39 for beta-diversity), indicating that TPOAb-seropositivity does not significantly differentiate gut microbiota composition and diversity. Interestingly, TPOAb status accounted for only a minor fraction (0.07%) of microbiome variance (p = 0.545). Further exploration of taxonomic differences identified 138 taxa nominally associated with TPOAb status. Among these, 13 taxa consistently demonstrated nominal significance across three additional DA methods, alongside notable associations within various functional pathways. Furthermore, we showed that ethnicity-specific reference intervals for serum thyroid biomarkers are not required, as no significant disparities in serum thyroid markers were found among the three ethnic groups residing in an iodine-replete area (p > 0.05 for thyrotropin, free thyroxine, and TPOAb).
Conclusion:
These findings suggest that there is no robust difference in gut microbiome between individuals with or without TPOAb in terms of alpha and beta-diversity. Nonetheless, several taxa were identified with nominal significance related to TPOAb presence. Further research is required to determine whether these changes indeed imply a higher risk of overt HT.
Introduction
Hashimoto's autoimmune thyroiditis (HT) is characterized by the progressive destruction of thyroid hormone-producing thyrocytes. It is the most common autoimmune endocrine disorder affecting ∼5% of the population in industrialized Europe and its prevalence has been steadily increasing worldwide, highlighting the impact of environmental factors. 1 The clinical onset of HT is preceded by high serum concentrations of thyroid peroxidase antibodies (TPOAb), which have been linked to an increased risk of developing thyroiditis. 2,3
The loss of immune self-tolerance to thyrocyte antigens is driven by a combination of genetic susceptibility and environmental exposures. The HT penetrance in monozygotic twins is concordant in only 55% of cases, 4 underscoring the importance of environmental factors in HT development, and genetics alone cannot explain the global increase in HT prevalence. Given its high prevalence and the knowledge gaps about specific environmental factors involved in HT onset, a better understanding of HT pathogenesis is needed. Since the intestinal microbiota plays an important role in the induction, training, and functioning of innate and adaptive immunity, 5 –8 alterations in the crosstalk between the gut microbiome and host immunity could be pivotal in autoimmune disease pathogenesis. 5,9 –11
Prior cross-sectional studies have reported perturbations in the microbiota composition of HT patients, characterized by reduced bacterial diversity, 12 –16 with correlations with clinical thyroid function parameters. 15,17 As those patients already had clinically significant hypothyroidism, it was unclear whether the microbial abnormality preceded the disease or vice versa. The nature of the microbiome structure before disease onset has remained unstudied. Moreover, the prior studies were conducted in small numbers of HT patients, including levothyroxine (LT4)-treated patients, and usually did not account for patient ethnicity and geography, which significantly affect gut microbiome composition, as shown in the multiethnic HEalthy Life In an Urban Setting (HELIUS) cohort. 18 However, serum thyroid antibody concentrations differ between ethnic groups 19 and persons of Western descent are more susceptible to autoimmune diseases than other ethnicities. 20,21
To address these issues, the primary aim of this study is to examine the relationship between gut microbiota composition and autoimmune thyroiditis in 1468 European Dutch, Moroccan, and Turkish participants who were clinically euthyroid, selected from the multiethnic HELIUS cohort. 22 Since these ethnic groups differ in the proportion of seropositivity and in the likelihood of developing clinical hypothyroidism, the secondary aim of the study is to discern whether these ethnic diversities warrant distinct reference intervals for accurately interpreting serum thyroid biomarkers.
Methods
Detailed descriptions of the methodology are listed in the Supplementary Methods section
The cross-sectional data used were obtained during baseline visits of the prospective multiethnic HELIUS cohort study. 22 All participants provided written informed consent and the study was approved by the Medical Ethics Review Board of the Amsterdam University Medical Center) (Amsterdam UMC), location AMC, and followed the principles of the Declaration of Helsinki (revisions 6 and 7). The Protocol ID is NL32251.018.10, with approval number 10/100# 10.17.1729. Based on the availability of fecal 16S rRNA sequencing data, a total of 1468 European Dutch, Moroccan, and Turkish participants ≥35 years old were included in this study. Excluding criteria are shown in Supplementary Figure S1.
Blood samples were collected during morning study visits under fasting conditions. Reference values ranged from 0.5 to 5.0 mU/L for thyrotropin (TSH) and 12–22 pmol/L for free thyroxine (fT4); TPOAb serum levels <30 kU/L were considered negative, 23 whereas TPOAb serum levels ≥60 mU/L were considered as positive.
Fecal bacterial compositions were profiled by sequencing the V4 region of the 16S rRNA gene on an Illumina MiSeq platform (Illumina RTA v1.17.28; MCS v2.5, San Diego, CA, USA). Statistical analyses were performed in the R statistical framework (v. 4.0.3; R Foundation for Statistical Computing, Vienna, Austria). The covariates included for all microbial analyses comprised age, sex, ethnicity, body mass index (BMI), and smoking status (“Yes currently” or “No”), in line with the specified methodology. 24 Multivariable linear regression was applied to assess the relationship between alpha-diversity indices and TPOAb presence. The Jaccard index, Bray–Curtis dissimilarity, Aitchison distance, and weighted and unweighted UniFrac distances were calculated for beta-diversity. Taxonomic differences were analyzed using MaAsLin2 as the primary differential abundance (DA) tool. These outcomes were verified by three additional DA tools: the Wilcoxon test of the CLR, ANCOM-BC, and ALDEx2.
Microbial data were summarized to phylum level. The four most abundant phyla were analyzed using MaAsLin2 (Microbiome Multivariable Associations with Linear Models) in R (package v. 1.8.0), 24 with age, sex, BMI, smoking, and ethnicity as covariates. LOG transformation, no normalization, the Linear Models analysis method, and no filter for abundance or prevalence were used as settings for MaAsLin2.
Results
Sex-specific, but comparable ethnic differences in thyroid marker distribution
The majority of all 1468 participants, in whom thyroid markers were analyzed, were of European Dutch descent (57.2%), followed by individuals of Moroccan (26.1%) and Turkish descent (16.8%). There were no differences in the distribution of concentrations of the serum thyroid markers, TSH, fT4, and TPOAb, between these three ethnic groups (Fig. 1). Serum TSH and TPOAb levels were significantly positively correlated with each other in European Dutch and Moroccan but not in Turkish participants (Fig. 2A); in contrast, serum TSH and fT4 levels showed inverse correlations in all three ethnic groups (Fig. 2B).
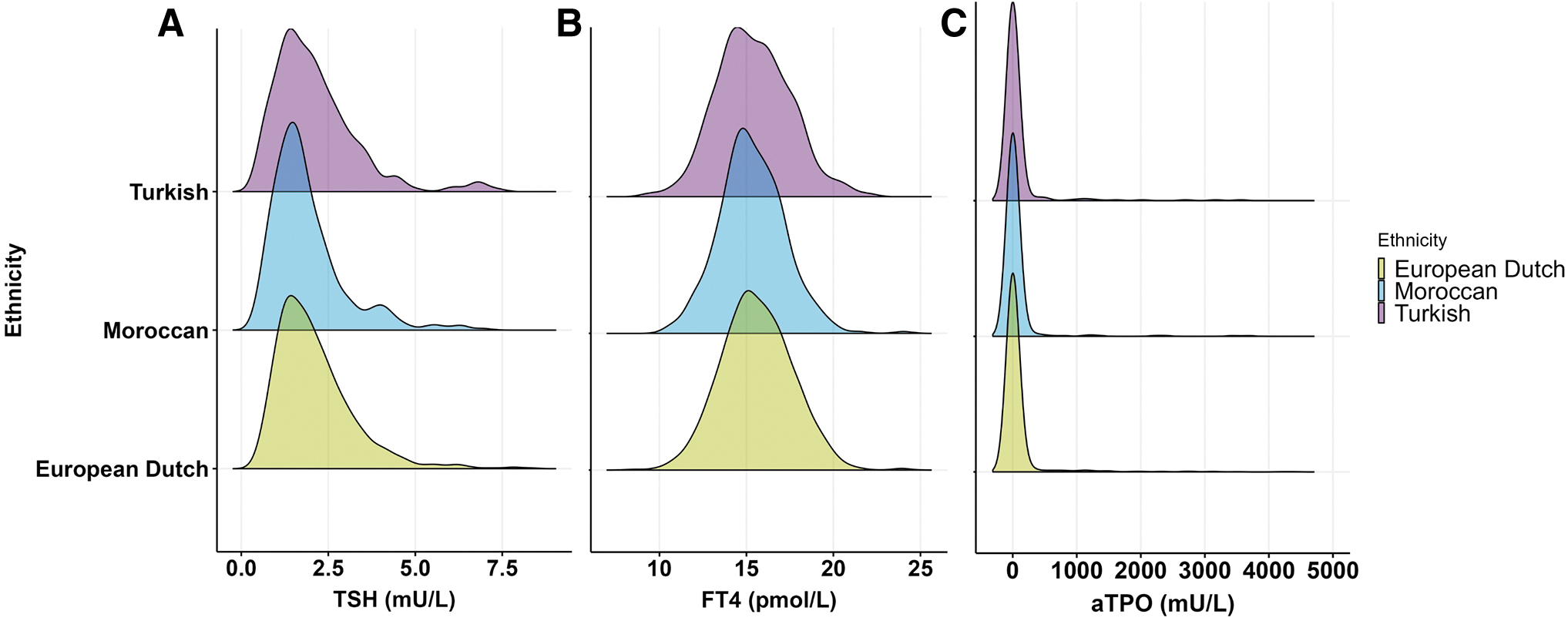
Thyroid markers are equally distributed among the 246 Turkish, 383 Moroccan, and 839 European Dutch participants. (
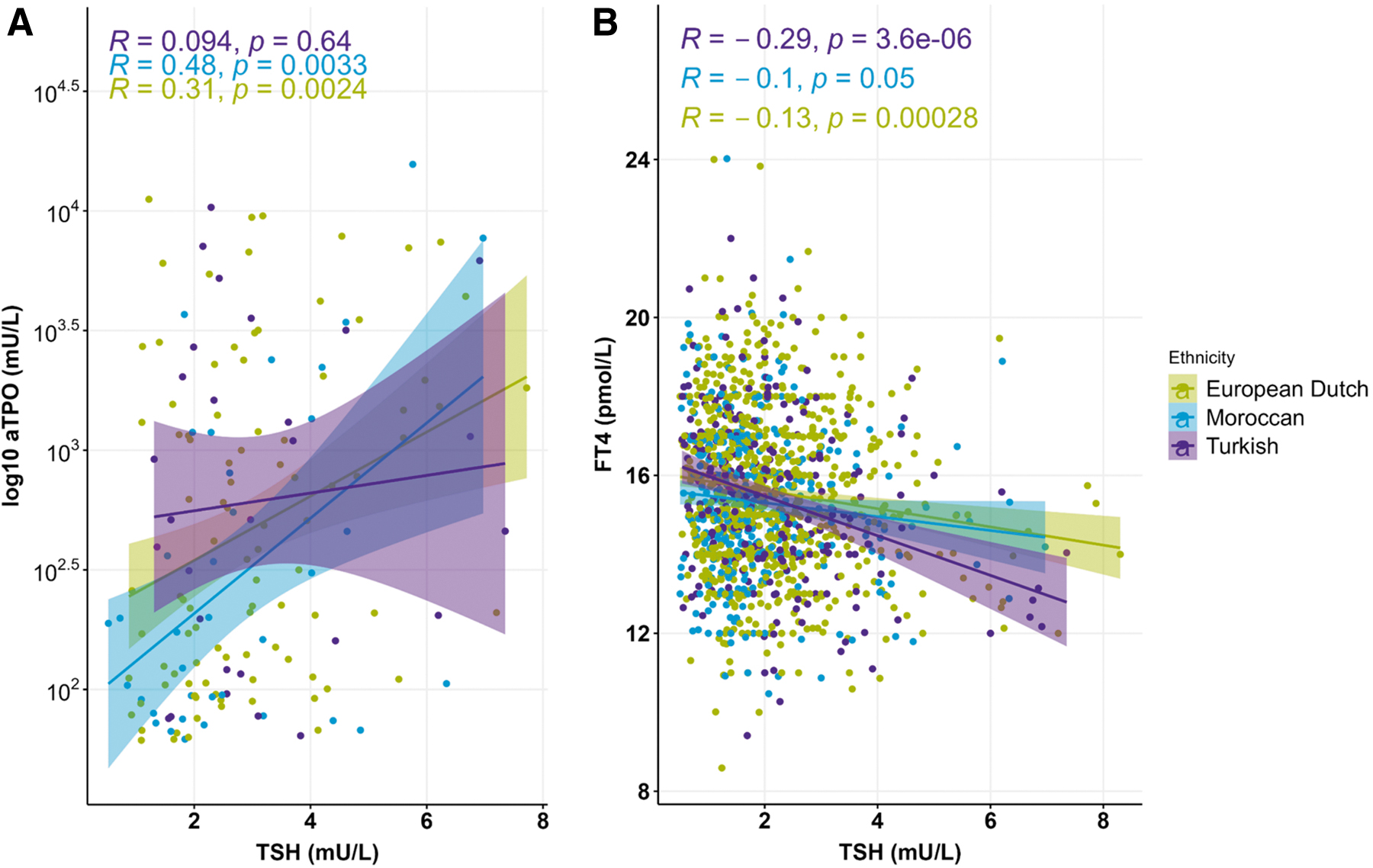
Significant correlation between the concentration of serum thyroid makers among the three different ethnicities. (
Of the 1468 participants, 1309 had no detectable serum levels of TPOAb (Table 1). As expected, 25,26 the prevalence of high TPOAb levels was greater in women than men (13.9% vs. 8.2%, p < 0.001). TPOAb-positive participants had significantly higher serum TSH (median 2.56 mU/L vs. 1.71 mU/L, respectively, p = < 0.001) and lower serum fT4 (mean 15.1 pmol/L vs. 15.5 pmol/L, respectively, p = 0.010) levels compared with TPOAb-negative individuals. No significant differences were found in anthropometric parameters, medication use, and metabolic profile between TPOAb-negative and TPOAb-positive participants. Similarly, dietary intake, 27 including total calories (kcal/day), carbohydrates (g/day), fiber (g/day), protein (g/day), and fat (g/day), was similar between the two groups.
Patient Characteristics of 1468 HEalthy Life In an Urban Setting Participants
For normally distributed parameters, data are presented as mean ± SD, and p-values were calculated using Student's t-test. For non-normally distributed parameters, data are presented as median [IQR], and the p-value was calculated using the Mann–Whitney U test. Nominal variables are presented as n (%).
Significant p-values are in bold.
BMI, body mass index; DBP, diastolic blood pressure; fT4, free thyroxine; HDL, high-density lipoprotein; IQR, interquartile range; LDL, low-density protein; SBP, systolic blood pressure; TPOAb, thyroid peroxidase antibodies; TSH, thyrotropin; WHR, waist–hip ratio.
TPOAb-seropositivity is not a differentiating factor of gut microbiota composition and diversity, but ethnicity is
There were no differences in alpha diversity of the fecal microbiota between the two groups in analyses with all six metrics used (Fig. 3A). Moreover, the microbiome population structure of seropositive and seronegative participants was overlapping; for example, analysis of Aitchison distance, a beta-diversity metric, showed no significant difference between the two groups (Fig. 3B), indicating that TPOAb presence per se is not a major contributing factor to gut microbial composition (Supplementary Table S1). Other indices of beta-diversity (Bray–Curtis index, Jaccard index, weighted and unweighted UniFrac distances) corroborated no significant difference between the seropositive and seronegative groups (Supplementary Fig. S2). In addition, none of the beta-diversity indices was significantly different for dispersion (data not shown), indicating homogeneous variation in the gut microbiome composition within each group. Thus, the overall ecological parameters of the two groups were indistinguishable in both composition and dispersion.
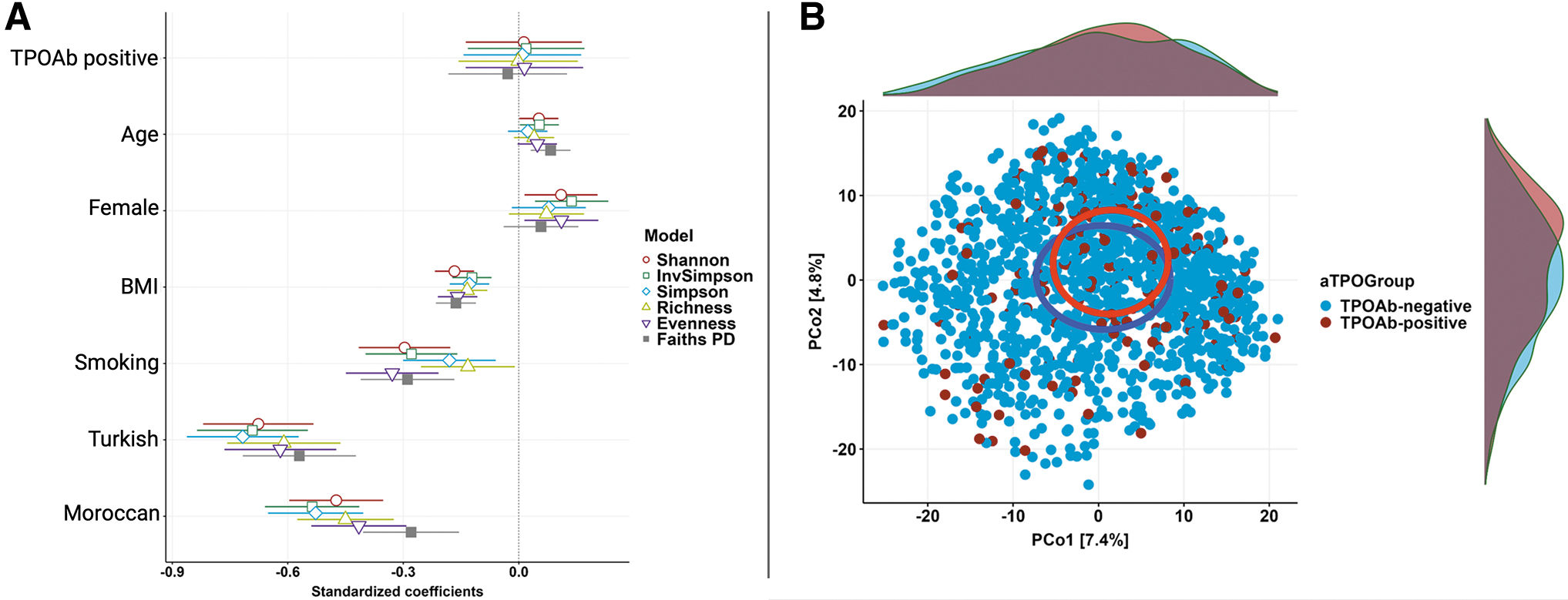
TPOAb presence is not a major contributing factor in gut microbiome diversity. (
Nevertheless, alpha- and beta-diversity analyses demonstrated that ethnicity is one of the most significant differentiating factors of gut microbial composition (2.3%, R 2 = 0.02, Supplementary Table S1).
Specific gut microbial taxa and functional pathways are significantly associated with TPOAb status
Applying the MaAsLin2 (Microbiome Multivariable Associations with Linear Models, software version 2) algorithm with confounder adjustment (age, sex, BMI, ethnicity, and smoking), we found 138 nominally significant taxa associated with TPOAb status (Supplementary Table S2, visualized at family level Fig. 4). A total of seven families were consistently and positively associated with the presence of TPOAb at the nominal p-value level of significance. The taxa with the highest effect-size coefficients belong to the Desulfovibrionaceae family, followed by Acidaminococcaceae, Mollicutes RF39, Flavobacteriales, Corynebacteriaceae, Defluviitaleaceae, and Coriobacteriaceae. In contrast, five taxa from three families had consistently negative associations with TPOAb presence (Erysipelotrichaceae and Clostridiales vadin BB60 group). The family with the highest negative coefficient was the Clostridiales vadin BB60 group. Three taxa of the Erysipelotrichaceae had a nominally significant association with TPOAb absence.
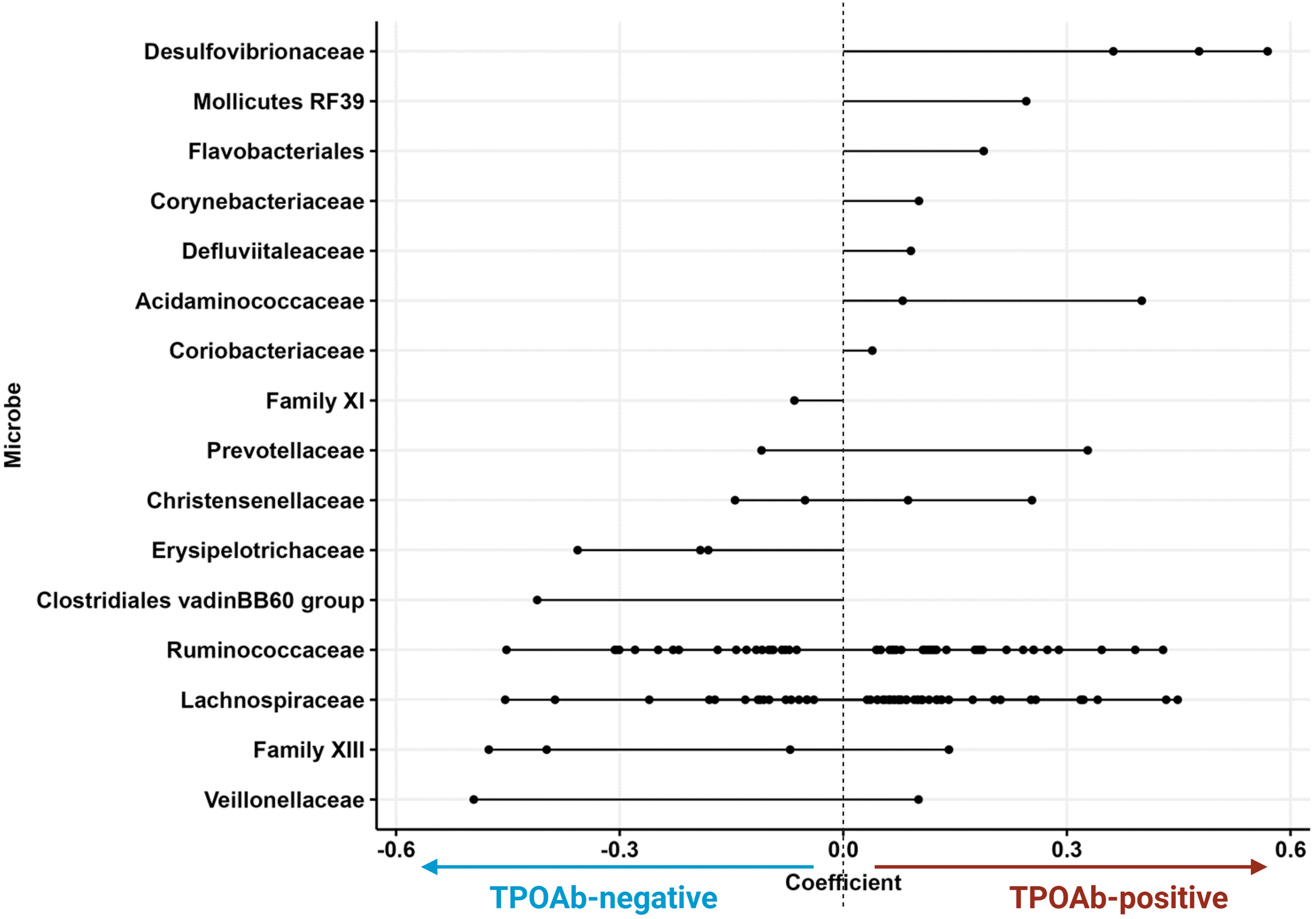
Microbial taxa associated with TPOAb status. Lollipop chart showing the discriminating families (or when family was not annotated; orders) between TPOAb-positive versus TPOAb-negative participants. Each dot represents a differentially significant abundant taxon (N = 138 individual taxa) at a certain phylogenetic level (ASV, genus, family), which is then grouped by the family level (each row). The direction of the line indicates the phenotype association (positive or negatively associated with TPOAb presence). The line length indicates the effect size (coefficient) with corresponding SE. Two significant lineages, Mollicutes RF39 and Flavobacteriales, are not annotated on the family level, and are shown on the order level. Exact p-values are stated in Supplementary Table S2. ASV, amplicon sequence variant; SE, standard error.
The families Prevotellaceae, Christensenellaceae, Ruminococcaceae, Lachnospiraceae, Family XIII, and Veillonellaceae consisted of multiple nominally significant taxa with abundances pointing in opposing directions. No taxa were significantly associated with TPOAb status after correcting for multiple testing using Benjamini–Hochberg q-values (adjusted p-value >0.05; Supplementary Table S2).
To substantiate the findings of MaAsLin2, we applied three additional DA tools (ALDEx2, CLR transformed Wilcoxon test, and ANCOM-BC). All methods showed similar results (Supplementary Table S3); 13 taxa were nominally significant in all 4 DA methods (Fig. 5A, B), but no significant changes in microbial abundances between the two groups were observed after adjusting for multiple testing.
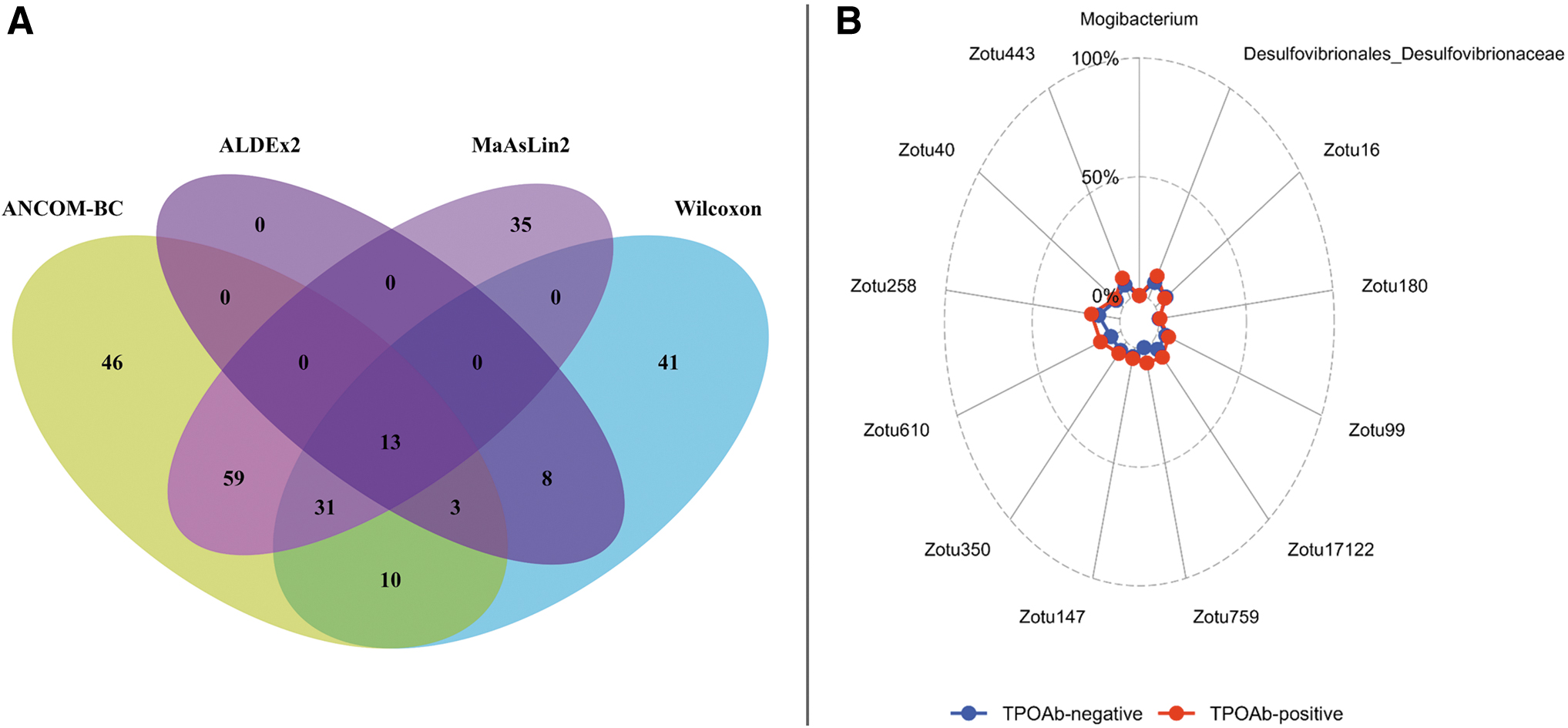
Comparing MaAsLin2 results with other DA tools identified 13 nominally significant overlapping taxa. (
Given the observed differences in TSH and fT4 levels between TPOAb-positive individuals and their control counterparts, potential correlations between these hormone levels and taxa abundance were explored. A total of 19 significant correlations were identified, with 4 taxa consistently found across all methods, including Zotu99 (Ruminococcaceae_UCG-013), Zotu258 (Marvinbryantia), Zotu40 (Blautia faeces) in relation to TSH, and Zotu759 (Ruminococcaceae_UCG-014) concerning fT4 (data not shown). Notably, all four taxa exhibited significant correlations with TPOAb status, as illustrated in Figure 5B.
The differential expression of microbial pathways was predicted by PICRUSt2, in participants with or without TPOAb. At the nominal significance level, multiple pathways were found to be differentially abundant between the TPOAb groups (Fig. 6 and Supplementary Table S4). Several pathways such as D-glucarate degradation (glucardeg PWY), lactose and galactose degradation (lactosecat PWY), glycol metabolism and degradation (glycol glyoxdeg PWY), glycolysis V, ethylmalonyl-CoA, and adenosylcobalamin biosynthesis I pathways were all decreased in TPOAb-positive participants. Three pathways were increased in TPOAb-positive participants, of which the mycolyl–arabinogalactan–peptidoglycan complex biosynthesis pathway (PWY 6397) had the highest effect size. No microbial pathway was significantly associated with TPOAb status after correcting for multiple tests using Benjamini–Hochberg q-values (adjusted p-value >0.05).
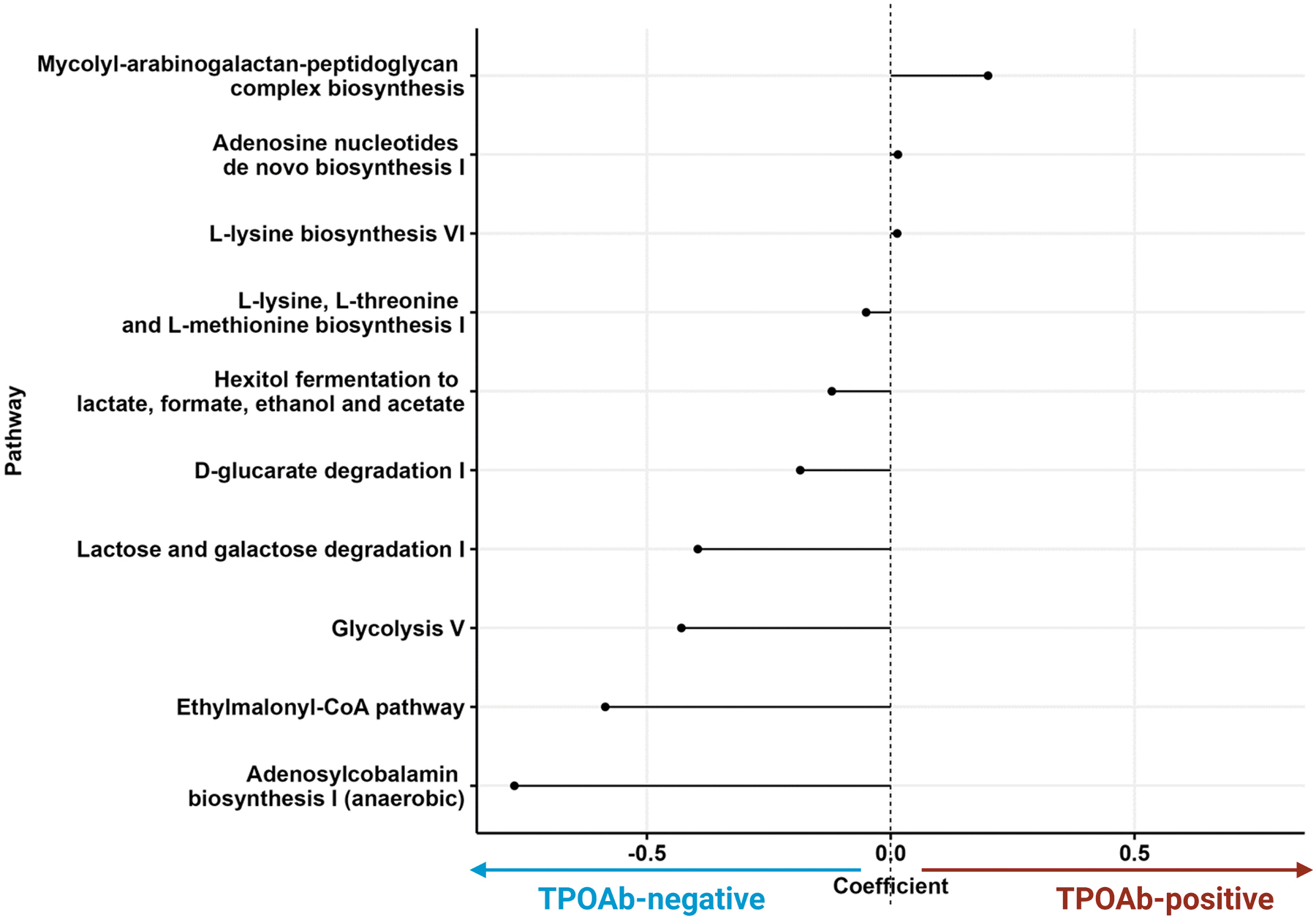
Microbial pathways associated with TPOAb status. Lollipop chart showing the discriminating microbially expressed pathways as imputed by PICRUSt2 between TPOAb-positive versus TPOAb-negative participants. Each dot represents one of the 10 differentially significant abundant pathways. The direction of the line indicates the phenotype association (3 positive and 7 negatively associated with TPOAb presence). The line length indicates the effect size (coefficient). p-Values for each pathway are in Supplementary Table S4.
Discussion
This study aimed to determine whether structural and/or functional gut microbiota perturbations occur in the early stages (seroconversion) of autoimmune thyroiditis. Given the lack of studies investigating the microbial pattern before disease onset, our study constitutes a step forward to better understanding the potential contribution of the gut microbiome in the pathogenesis of autoimmune thyroiditis. The main results from our large multiethnic population-based cohort study suggest that euthyroid persons with TPOAb (before the clinical onset of autoimmune thyroid disease) exhibit only modest gut microbial variation at the taxonomic level compared with those without these antibodies, whereas no significant differences in global alpha and beta diversity were found (Supplementary Fig. S3). Future research is needed to effectively evaluate whether new gut microbiome-based diagnostics, preventives, or therapeutics for autoimmune hypothyroidism would reduce the need for the current replacement with daily oral administration of synthetic thyroid hormone (LT4) after the thyroid gland has sustained substantial damage.
Similar to prior cohort studies in adults, 23,25 10.8% of the 1468 euthyroid participants in the HELIUS cohort had TPOAb serum antibodies. The observed difference in the proportion of thyroid peroxidase (TPO)-positivity between men and women reflects the well-known sex difference, as HT is four to eight times more common in women than in men. 25,26 Variance in biomarker values has been noted between different ethnic groups, 28 highlighting the importance of ethnicity-specific reference intervals for biomarkers. Our study did not find any differences in serum thyroid markers or thyroid autoimmunity between these three ethnic groups living in an iodine-replete area. However, consistent with prior analyses of multiethnic population studies, 18 we show that ethnicity is an important correlate with microbiome diversity.
A growing body of evidence has shown that gut microbiota dysbiosis may be a marker of several autoimmune diseases, including type 1 diabetes (T1D), inflammatory bowel disease, and rheumatoid arthritis. 6,29 –31 Yet, studies on the association between autoimmune thyroiditis and gut microbiome composition have been limited and have yielded conflicting results. 11,13 –17,32 –34 This may reflect small sample sizes with heterogeneous groups and varying definitions of autoimmune thyroiditis. 34 For example, two studies investigated the differences in intestinal microbiota in hypothyroid participants without detectable TPOAb serum levels, 13,32 thus including nonautoimmune causes of hypothyroidism. The conflicting outcomes observed in prior studies may reflect thyroid status differences of the subjects. Whereas some studies included only euthyroid HT patients using LT4, 16,17 others included both euthyroid and hypothyroid HT patients, 13 –15 or solely included hypothyroid (medication-naive) patients with autoimmune thyroiditis. 32,33 Such methodological differences make comparison of results difficult; for example, the impact of LT4 use on gut microbiota composition is currently unknown.
Many of these studies were conducted in China 13,17,32,33 and might not generalize to other populations as ethnicity and patient geography are important covariates of the gut microbiome composition. 18 In conclusion, it remains difficult, if not impossible, to validate our results with previously published cohorts.
The fecal taxa most predictive of TPOAb-positive status belonged to the family Desulfovibrionaceae and was found nominally significant in analyses with all four DA tools. While prior studies on the gut microbiome composition of patients with HT did not report on the abundance of the Desulfovibrionaceae family, higher abundances of Desulfovibrio, a genus within this family, have been inversely associated with other autoimmune diseases such as T1D; 35 the presence of Desulfovibrio piger in duodenal biopsies predicted preserved beta-cell function in T1D patients, consistent with a protective effect. 35
Several taxa within the same family (e.g., the families Prevotellaceae, Christensenellaceae, Family XIII, Ruminococceae, Lachnospiraceae) had bidirectional associations with TPOAb presence. These findings reflect the heterogeneity of these taxonomic families, which substantially vary in genetic content and, thus function. Considering the large effect size and high average abundance of two taxa belonging to the same family (e.g., two Veillonellaceae taxa), those might represent two separate species having opposing effects on TPOAb pathogenesis. Our imputation of pathways using PICRUSt2 did not permit discrimination between the genetic functions of these microbes. Accordingly, differences within major taxa might also explain the discordances found in prior studies 11,13 –17,32,33 but do not rule out a role in HT pathogenesis.
Functional profiling of gut microbial pathways showed a decrease in the D-glucarate degradation pathway in the TPOAb-positive participants. 36,37 D-Glucaric acid is the end product of D-glucarate degradation, serving as an energy source for certain bacteria, 36 and is a potent β-glucuronidase inhibitor. 37 β-glucuronidase promotes the reabsorption of free thyroid hormones into the enterohepatic circulation by hydrolyzing conjugated iodothyronines. 38,39 It is unknown whether this observed decrease in the D-glucarate degradation pathway affects (conjugated) iodothyronine concentrations. In conclusion, the differently abundant families and functional pathways identified in this study do not indicate a priori immunoregulatory function, thus their significance is uncertain.
Strengths and limitations of the study
This study has several strengths, including its large sample size, inclusion of three ethnicities, and use of the same analytical tests and study instruments across the population. All study participants were euthyroid and did not use LT4 at the time of fecal sampling, leading to a highly homogeneous cohort without the confounding effect of thyroid treatment. This is the first study to assess the gut microbiome composition in euthyroid TPOAb-positive patients who did not have overt clinical autoimmune thyroid disease. Another notable strength is that we addressed potential confounders since both intrinsic and extrinsic factors modulate gut microbiota composition. Study participants with recent antibiotic use were excluded, and we adjusted our analyses for other relevant confounding factors. Lastly, at the compositional level (alpha- and beta-diversity) and for the DA results, we applied multiple indices, dissimilarity metrics, and methods to substantiate our findings.
One study limitation is its cross-sectional design. To understand the drivers of the gut microbiome profile, it is necessary to follow participants prospectively. Although ∼10% of the general population is positive for TPOAb, fortunately, not all will develop overt hypothyroidism. 40 The natural course of autoimmune hypothyroidism is marked by slow development over years, evolving from positive serum levels TPOAb with serum thyroid hormone levels remaining within the reference range (euthyroidism), evolving to subclinical and eventually overt hypothyroidism. The risk of developing overt hypothyroidism over 13 years for euthyroid TPOAb-positive women was 55.2% (37.1–73.3%). 41 Longitudinal data are thus needed to investigate whether the differences in microbiome composition are indeed related to progression to overt hypothyroidism. Such longitudinal cohorts also will be better suited for studying the effect of LT4 supplementation on microbiome composition.
As this is a hypothesis-generating study, we report results at the nominal significance level; however, this means that the results may be false-positive findings, and all associations require validation in independent cohorts. Currently, multiple DA tools are available, and they are frequently used interchangeably, resulting in inconsistent findings that may hinder comparability and reproducibility across studies. 29 To address this issue, a consensus approach or standardized guideline is necessary for analyzing complex microbiome data. Hence, we employed multiple DA methods and focused on significant features identified by most tools to increase our results' robustness.
Furthermore, the microbial functional pathways were imputed using PICRUSt2, which is based on reference genomes for the 16S gene region sequenced; this is less accurate than shotgun metagenomic sequencing, which provides the actual genetic profile of the fecal microbiome. 42 Finally, the distribution of TPOAb is skewed toward zero, as 1309 participants showed no detectable anti-TPO antibodies. Despite reporting the largest cohort of HT participants, this study still has low statistical power due to the high heterogeneity of the human gut microbiome and the relatively low incidence of TPOAb in the population.
Conclusions
Neither by global ecological patterns nor by microbial taxa or functional pathway level did we find robust evidence of gut microbiome differences in relation to TPOAb status. However, we report several nominally significant taxa associated with TPOAb presence, 13 concordant across four DA methods. Future clinical prospective studies and translational animal investigations are warranted to elucidate the potential contribution of the identified taxa concerning susceptibility to HT onset and/or accelerating disease progression.
Footnotes
Acknowledgments
The authors are most grateful to the participants of the HELIUS study and the management team, research nurses, interviewers, research assistants, and other staff who have taken part in gathering the data for this study.
Authors' Contributions
Conceptualization, supervision, project administration: M.N. Methodology, software, formal analysis, visualization, and writing—original draft: A.C.F. and U.B. Resources: B.-J.H.v.d.B. and H.G. Data curation: U.B and H.G. Writing—review and editing: A.C.F., U.B., D.C., A.H.v.d.S., E.R, B.-J.H.v.d.B., H.G., A.H.Z., E.F., M.J.B., and M.N. Funding acquisition: B.-J.H.v.d.B and M.N.
Author Disclosure Statement
M.N. is on the Scientific Advisory Board of Caelus Pharmaceuticals, the Netherlands. None of these is directly relevant to the current article. There are no patents, products in development, or marketed products to declare. The other authors declare no competing financial interests. A.C.F., U.B., D.C., H.G., A.H.Z., B-J.H.v.d.B., E.R., A.H.v.d.S., E.F., and M.J.B. have no conflicts to disclose.
Funding Information
A.C.F. is appointed on a LeDucq Foundation TransAtlantic consortium grant 2017 17CVD01 (to
Supplementary Material
Supplementary Methods
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4