
Abstract
Introduction:
Thyroid hormone transporters are essential for thyroid hormones to enter target cells. Monocarboxylate transporter (MCT) 8 is a key transporter and is expressed at the blood–brain barrier (BBB), in neural cells and many other tissues. Patients with MCT8 deficiency have severe neurodevelopmental delays because of cerebral hypothyroidism and chronic sequelae of peripheral thyrotoxicosis. The T3 analog 3,3′,5-triiodothyroacetic acid (TRIAC) rescued neurodevelopmental features in animal models mimicking MCT8 deficiency and improved key metabolic features in patients with MCT8 deficiency. However, the identity of the transporter(s) that facilitate TRIAC transport are unknown. Here, we screened candidate transporters that are expressed at the human BBB and/or brain–cerebrospinal fluid barrier and known thyroid hormone transporters for TRIAC transport.
Materials and Methods:
Plasma membrane expression was determined by cell surface biotinylation assays. Intracellular accumulation of 1 nM TRIAC was assessed in COS-1 cells expressing candidate transporters in Dulbecco’s phosphate-buffered saline (DPBS)/0.1% glucose or Dulbecco’s modified Eagle’s medium (DMEM) with or without 0.1% bovine serum albumin (BSA). Expression of Slc22a8 was determined by fluorescent in situ hybridization in brain sections from wild-type and Mct8/Oatp1c1 knockout mice at postnatal days 12, 21, and 120.
Results:
In total, 59 plasma membrane transporters were selected for screening of TRIAC accumulation (n = 40 based on expression at the human BBB and/or brain–cerebrospinal fluid barrier and having small organic molecules as substrates; n = 19 known thyroid hormone transporters). Screening of the selected transporter panel showed that 18 transporters facilitated significant intracellular accumulation of TRIAC in DPBS/0.1% glucose or DMEM in the absence of BSA. In the presence of BSA, substantial transport was noted for SLCO1B1 and SLC22A8 (in DPBS/0.1% glucose and DMEM) and SLC10A1, SLC22A6, and SLC22A24 (in DMEM). The zebrafish and mouse orthologs of these transporters similarly facilitated intracellular accumulation of TRIAC. Highest Slc22a8 mRNA expression was detected in mouse brain capillary endothelial cells and choroid plexus epithelial cells at early postnatal time points, but was reduced at P120.
Conclusions:
Human SLC10A1, SLCO1B1, SLC22A6, SLC22A8, and SLC22A24 as well as their mouse and zebrafish orthologs are efficient TRIAC transporters. These findings contribute to the understanding of TRIAC treatment in patients with MCT8 deficiency and animal models thereof.
Introduction
Thyroid hormones are crucial for development, particularly of the brain. The biological availability of thyroid hormone in the brain is dependent on thyroid hormone cell membrane transporter proteins at the blood–brain barrier (BBB) and the blood–cerebrospinal fluid barrier (BCSFB), dependent on the developmental stage. 1 Monocarboxylate transporter 8 (MCT8) encoded by solute carrier (SLC)16A2, which is expressed at the BBB and in neural cells, is crucial for transport of 3,3′,5-triiodothyronine (T3) and thyroxine (T4). 2 –4 Patients with MCT8 deficiency have severe neurodevelopmental delays because of cerebral hypothyroidism and suffer from chronic sequelae of peripheral thyrotoxicosis. 5
Preclinical studies indicated the potential value of the T3 analog 3,3′,5-triiodothyroacetic acid (TRIAC). TRIAC can enter cells independent of MCT8, is metabolized by the type 3 deiodinase (D3) similar to T3 and is even a better substrate for the type 1 deiodinase (D1). In addition, TRIAC is able to induce similar expression of thyroid hormone responsive genes as T3. 6 In Mct8/Oatp1c1 double knockout (DKO) mice, which have neurodevelopmental abnormalities because of impaired thyroid hormone transport into CNS, early postnatal administration of TRIAC restored cerebellar Purkinje cell dendritogenesis and cortical myelination, improved white matter thinning, brain network dysfunction and locomotor performance. 6 –8 Similar beneficial effects on brain development were observed in TRIAC-treated mct8 deficient zebrafish or chicken eggs. 9,10 In patients with MCT8 deficiency, TRIAC treatment improved various metabolic and cardiovascular outcomes. 11,12 The effects of early TRIAC intervention on neurocognitive outcomes in children with MCT8 deficiency are currently being investigated (NCT02396459). On the basis of these lines of evidence, TRIAC represents a promising candidate for treatment for MCT8 deficiency. However, it is unclear which transporter(s) facilitate TRIAC transport into the brain and other tissues. In this study, we screened candidate transporters and known thyroid hormone transporters to identify TRIAC transporters.
Materials and Methods
Reagents
TRIAC (purity ≥ 90%) was purchased from Sigma Aldrich (Zwijndrecht, the Netherlands). [125I]-TRIAC was prepared as previously described. 13 Dulbecco’s phosphate-buffered saline (DPBS) and Dulbecco’s modified Eagle medium/nutrient mixture F-12 (DMEM/F12) (HEPES, no phenol red) (abbreviated as DMEM) were purchased from Thermo Fisher (Leiden, the Netherlands).
Selection of candidate TRIAC transporters
The following strategy was used to shortlist candidate TRIAC transporters. First, all known transporters that facilitate transport of (sulfated) thyroid hormones were selected. Next, the Uniprot knowledge database was utilized, and publications were screened to identify potential transporters from the SLC and ABC cassette families that use substrates (monocarboxylates, amino acids, and oligopeptides, organic anions, sulfated and glucuronidated small substrates, nucleotides, and small cofactors), which may increase the likelihood for TRIAC transport. This list of putative transporters was further limited to those that have a relevant expression at the human BBB and/or the BCSFB, based on transcriptome analysis including human primary brain endothelial cells, induced pluripotent stem cell (iPSC)-derived brain endothelial cells, human choroid plexus epithelium, the human brain endothelial cell model hCMEC/D3, and the choroid plexus epithelial cell line HIBCPP (Supplementary table S1). 14 –17 Ultimately, 59 transporters (n = 40 based on their expression in at the human BBB and/or BCSFB; n = 19 known thyroid hormone transporters) were selected as candidates to screen for TRIAC transport.
Patients and placentas
The study received an exemption for approval from the Medical Ethics Review Committee of Erasmus MC according to the Dutch Medical Research with Human Subjects Law (MEC-2017-418). All patients provided written consent before donating their placentas. Healthy placentas of uncomplicated singleton pregnancies were collected immediately after delivery (via cesarean section) at Erasmus University Medical Center, Rotterdam, the Netherlands. Placentas with maternal viral infections (human immunodeficiency virus [HIV], hepatitis B, Zika and severe acute respiratory syndrome coronavirus 2 [SARS-CoV-2]), maternal diabetes, or fetal congenital abnormalities observed on ultrasound were excluded.
Transporter constructs
The constructs used in this study are described in the Supplementary material. Briefly, most constructs of the known thyroid hormone transporters and μ-crystallin (CRYM) were generated in previous studies (Supplementary table S2). We also included the organic anion transporters (OATs) in SLC22 family that were recently identified as (sulfated) thyroid hormone transporters, namely SLC22A8, SLC22A9, SLC22A11, and SLC22A24. 18 We also included SLC22A6 as the zebrafish orthologs of SLC22A6/8 are (sulfated) TH transporters. 18 The SLCO2B1 construct was generated as described in the Supplementary material. The SLC candidate transporters and thyroid hormone transporters OATP3A1 and OATP4C1 in pDONR vector were gifts from RESOLUTE Consortium & Giulio Superti-Furga (Addgene plasmids) and subcloned into pcDNA3.2-DEST-V5 vector (Thermo Fisher) via a cloning kit Gateway™ LR Clonase™ II Enzyme mix (Thermo Fisher) (Supplementary table S3). The ATP-binding cassette (ABC) transporters in mammalian expression constructs, containing a C-terminal DYKDDDDK tag, were purchased from GenScript (Leiden, the Netherlands). The commercial ABCC2 construct contained a missense mutation (Try39Phe), which was corrected to wild type (WT) using QuickChange Site-Directed Mutagenesis Kit (Agilent, Middelburg, the Netherlands) and specific primers (forward: 5′-caggagccataggtagcccaagggaatccac-3′; reverse: 5′-gtggattcccttgggctacctatggctcctg-3′) (Supplementary table S4). The mouse and zebrafish Slc10a1 constructs were purchased from GenScript and mouse Slco2b1 from GeneCopoeia (Rockville, USA) (Supplementary table S5). The sequences of the gene inserts were confirmed by Sanger sequencing (Eurofins, Ebersberg, Germany) by comparing them to the reference sequences from National Center for Biotechnology Information (NCBI).
Cell culture and transfection
COS1 cells (African green monkey kidney fibroblast-like cell line) (CVCL_0223) were purchased from ECACC (Sigma–Aldrich, Zwijndrecht, The Netherlands) and cultured as described before. 19 As previously reported, COS1 cells showed negligible T3 and T4 conversion indicating no relevant activity of the deiodinases. 20 This characteristic makes COS1 cells a suitable cell line for thyroid hormone transport studies. COS1 cells were transfected with the transporter plasmids as described previously. 21,22 For SLC7A11, the same amount of plasmid of the heavy chain hSLC3A2 (CD98), which is required for its activity, 23 was co-transfected. All cells were co-transfected with the intracellular iodothyronine binding protein CRYM, which also binds TRIAC, thereby minimizing TRIAC efflux and TRIAC being catalyzed by other (unknown) metabolic pathways than the deiodinases and thus potentiating intracellular accumulation. 24
Intracellular accumulation experiments
As a general screening strategy for TRIAC transport, we used two different media conditions, which are DPBS/0.1% glucose (amino acid free) and DMEM (amino acid rich). Among the selected candidate transporters, some of them can transport amino acids, such as SLC7A5, SLC7A11, and SLC16A10. 25,26 By using DPBS/0.1% glucose medium, we can minimize the effect of amino acids competing with TRIAC transport. On the contrary, transporters such as some SLC22s may need counter substrates for exchanging substrates. 27 Therefore, our strategy comprising two media aimed to minimize the chances of missing relevant transporters. Intracellular accumulation experiments of 1 nM 125I-TRIAC (50,000 counts per minute) were performed in transfected COS1 cells as described previously. 21,22 Cells were first briefly rinsed with incubation medium (DPBS/0.1% glucose or DMEM) and then incubated in the incubation medium in the absence or presence of 0.1% bovine serum albumin (BSA). As TRIAC binds to albumin, 28 we used BSA-free media, in which the free TRIAC concentration is equal to the added TRIAC concentration in the media, and media containing 0.1% BSA to represent a more physiological condition in which the substrate is bound to serum distributor proteins. Under the latter condition, the free TRIAC concentration is expected to be in the picomolar range and is continuously provided by release from BSA when TRIAC is taken up by the cells. 2 The incubation medium and incubation times are indicated in the figure legends. After incubation, cells were briefly washed with incubation medium in the presence of 0.1% BSA and lysed with 0.1 N NaOH. The intracellular radioactivity was measured in a gamma counter. Data are expressed as % net intracellular TRIAC, which was calculated as the % intracellular 125I-TRIAC in cells expressing transporters minus the % intracellular 125I-TRIAC in empty vector (EV) control cells.
Animals
Animal studies were executed in accordance with the European Union (EU) directive 2010/63/EU on the protection of animals used for scientific purposes and in compliance with the local regulations by the Animal Welfare Committee of the Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen (LANUV, Recklinghausen, Germany). Mct8/Oatp1c1 DKO mice (on C57BL/6 background) were generated by intercrossing MCT8 KO (MGI: 3710233) and OATP1C1 KO mice (MGI: 5308446) and genotyped as described elsewhere. 29 Mice were kept in individually ventilated (IVC) cages in the central animal facility of the University Hospital Essen.
Fluorescence in situ hybridization
Brain mRNA expression of Slc22a8 and Pecam1 (Cd31) was analyzed in male WT and Mct8/Oatp1c1 DKO mice by hybridization chain reaction fluorescent in situ hybridization (FISH). WT and DKO mice at postnatal days P12, P21, and P120 were killed by decapitation, brains were removed and frozen in 2-methylbutane cooled on dry ice. Coronal brain sections of three mice per genotype and time point were cut on a cryostat with a thickness of 20 µm. Sections were fixed with 4% paraformaldehyde/PBS for 1 hour, permeabilized in 0.4% Triton X-100 for 10 minutes and dehydrated with increasing concentrations of ethanol (50%, 70%, and 100%). Sets of 20 split-initiator probe pairs for detection of mouse Slc22a8 (NM_001164634.1) and mouse Pecam1 (NM_008816.3) were obtained from Molecular Instruments, Inc. (Los Angeles, USA). Hybridization was carried out according to published protocols. 30 Slc22a8 signals were amplified using B1-amplifier coupled to 647 fluorophore and Pecam1/Cd31 signals were amplified using the B4-546 amplifier. Upon posthybridization, sections were counterstained with DAPI and fluorescence images were taken in cortical and choroid plexus regions using a Leica SP8 confocal microscope (Leica Microsystems) and the Leica Application Suite X Software. The images were adjusted for brightness and contrast using ImageJ 1.54.
Quantitative real-time polymerase chain reaction
For quantifying Slc22a8 and Pecam1 (CD31) mRNA expression in different brain areas, total RNA was purified from isolated cerebral cortices and hippocampi of P12, P21, and P120 WT mice (5–7 animals/age group) with the NucleoSpin RNA Plus Kit (Macherey-Nagel, Dueren, Germany). cDNA was synthesized with the help of qScript cDNA Supermix (Qantabio, Beverly, USA), and quantitative real-time polymerase chain reaction (qRT-PCR) was performed on the CFX Opus 96 detection system (Bio-Rad, Feldkirchen, Germany) using Perfecta SYBR Green Supermix (Qantabio). The Slc22a8-specific primer pair (TGGCTACCTTCAACGGCAAG and CAGGATGGACACTCGGAACAA) and CD31 primers (AGCCTAGTGTGGAAGCCAAC and GCTCAAGGGAGGACACTTCC) were used. Transcript levels of the housekeeping gene cyclophilin D were assessed with the primer pair: GCAAGGATGGCA-AGGATTGA and AGCAATTCTGCCTGGATAGC. The relative gene expression was calculated with delta-delta Ct method.
Statistical analysis
The results of transport studies are presented as mean ± SEM of 3–6 independent experiments performed in two technical duplicates. The statistical analysis was performed using GraphPad Prism version 8.4.0. One-way ANOVA followed by Dunnett’s multiple comparisons test was used for statistical analysis. The data of the qRT-PCR experiments are represented as mean ± standard deviation. One-way ANOVA followed by Sidak’s multiple comparison post hoc test was conducted with GraphPad Prism 9.5. Differences were considered significant with p < 0.05.
Results
Screening for TRIAC transporters
First, using COS1 cells expressing these transporters, we performed cell surface biotinylation assays to assess transporter expression at the plasma membrane. Most transporters were robustly expressed at the plasma membrane, except for SLC1A4, SLC36A4, SLC52A2, SLC52A3, ABCC1, and ABCC10 which were not or hardly detectable (Supplementary Fig. S1 and S2). Therefore, these transporters were excluded from further analysis.
To test TRIAC transport, we incubated transfected cells with 1 nM radiolabeled TRIAC and measured radioactivity in cell lysates. For incubation medium, we either used DPBS supplemented with 0.1% glucose as a plain medium or DMEM as a rich medium to provide potential counter substrates should there be transporters acting as exchangers.
In DPBS/0.1% glucose medium, the following transporters induced mild (<1.5 times over EV control) but statistically significant intracellular TRIAC accumulation: SLC15A1, SLC15A2, SLC38A1, SLC38A2, and ABCG4 as well as the known thyroid hormone transporters SLC10A1, SLCO1B1, SLCO2B1, SLC22A6, SLC22A8, SLC22A9, and SLC22A24 (Fig. 1A, B and C). In DMEM medium, the following transporters induced mild (<1.5 times over EV control) intracellular TRIAC accumulation: SLC7A11, SLC13A3, SLC15A2, SLC16A5, SLC38A1, SLC38A2, ABCC5, and ABCG4 as well as some known thyroid hormone transporters SLC10A1, SLC16A2, SLC16A10, SLCO1B1, SLC22A6, SLC22A8, and SLC22A9 (Fig. 2A, B and C). The latter group showed a potentiated accumulation in DMEM versus DPBS/0.1% glucose medium. For the 40 transporters that are not known to transport thyroid hormone, we also tested T3 and T4 transport in transfected COS1 cells. Only ABCG1 showed marginal induction of intracellular T3 accumulation in DPBS/0.1%nglucose medium (1.1 times over EV control), and ABCG5 induced intracellular T4 accumulation in DMEM medium (1.7 times over EV control) (Supplementary Figs. S3 and S4).
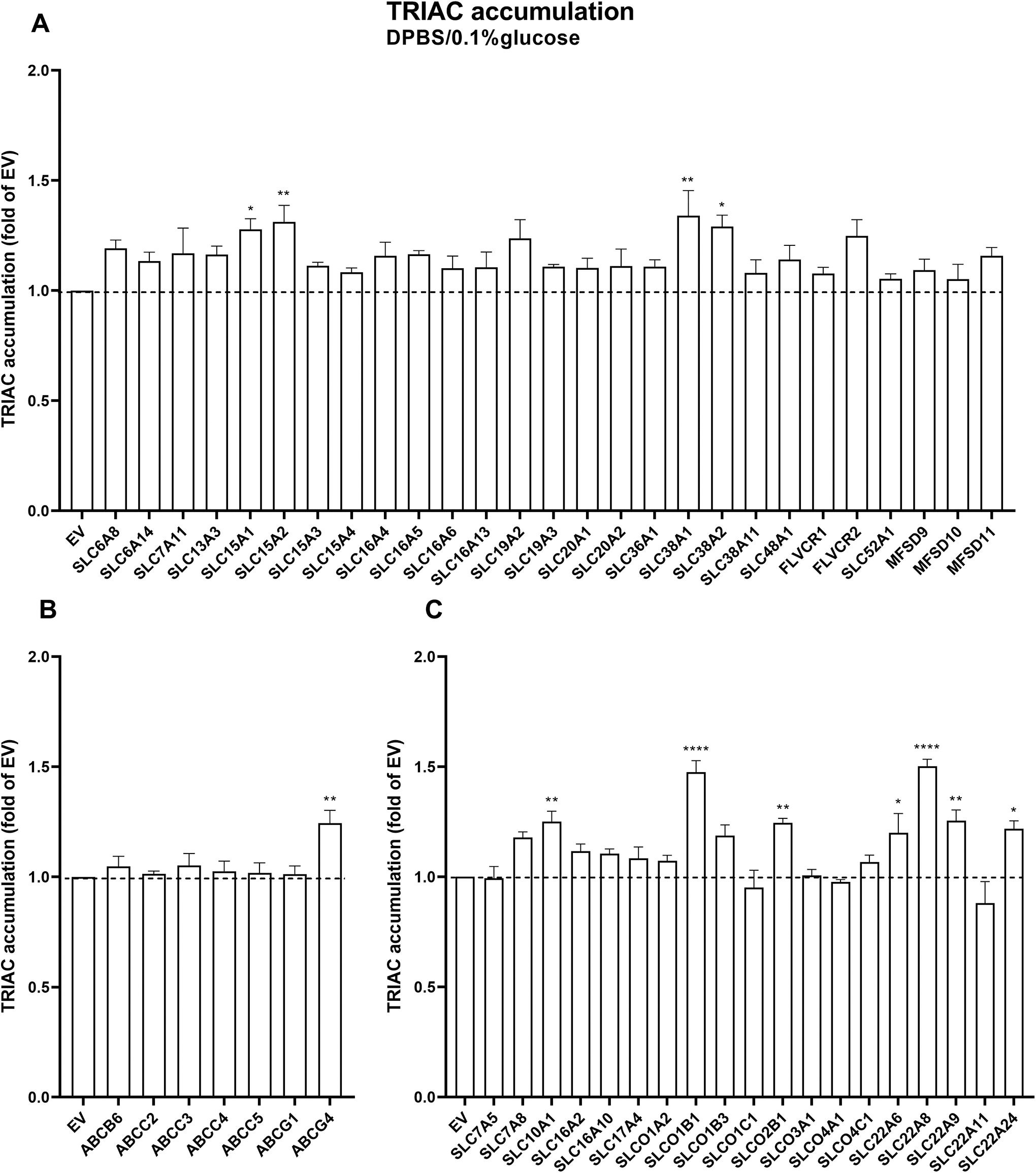
TRIAC accumulation in COS1 cells co-expressing CRYM and SLC

TRIAC accumulation in COS1 cells co-expressing CRYM and SLC
Next, we selected transporters that induced TRIAC accumulation in cells in either incubation medium and tested TRIAC accumulation in the presence of 0.1% bovine serum albumin (BSA). Cells expressing SLCO1B1 and SLC22A8 showed significant intracellular TRIAC accumulation when tested with both incubation media, whereas cells expressing SLC10A1, SLC22A6 and SLC22A24 also showed significant intracellular TRIAC accumulation in DMEM/0.1%BSA medium (Fig. 3A and B). Furthermore, TRIAC accumulation for these five transporters increased within 60 minutes incubation time, indicating that CRYM binding capacity was not saturated during the incubation time we selected for screening (Fig. 3C).
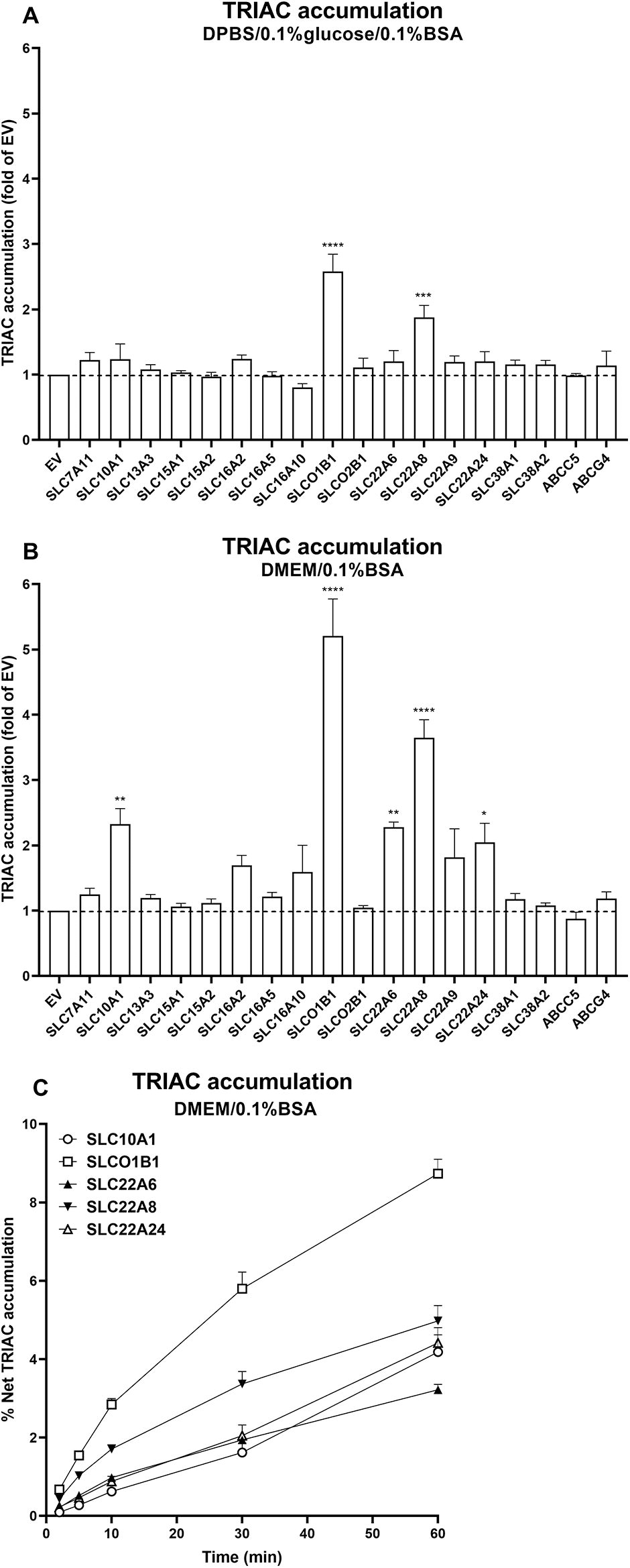
TRIAC accumulation in COS1 cells co-expressing CRYM and SLC or ABC transporters. 1 nM TRIAC accumulation experiments were performed in DPBS/0.1%glucose/0.1%BSA
To validate our findings in another cell line, we tested intracellular TRIAC accumulation in DMEM/0.1% BSA in HeLa cells transfected with these transporters. This showed a similar pattern of induction as in the COS1 cells, although not all of them reached statistical significance (Supplementary Fig. S5A, B and C).
Intracellular accumulation of TRIAC by zebrafish and mouse orthologs
As mouse and zebrafish models have been widely used to investigate the therapeutic potential of TRIAC for MCT8 deficiency, we also tested the mouse and zebrafish orthologs of the five human TRIAC transporters. Among these, SLC22A24 neither has mouse nor zebrafish orthologs, whereas SLCO1B1 only has a mouse ortholog SLCO1B2. DrOatx and drSlc22a6l have been reported to be the zebrafish orthologs of SLC22A6/8. 31 In transfected COS1 cells, all the mouse and zebrafish orthologs induced significant intracellular TRIAC accumulation in DPBS/0.1% glucose/0.1% BSA (two times over EV control, except mmSLCO1B2, which was 1.7 times) (Fig. 4A). In DMEM/0.1% BSA, all the orthologs achieved >2 times intracellular TRIAC uptake over EV control except mmSLC10A1 (1.6 times) (Fig. 4B). In transfected HeLa cells, all the orthologs induced significant intracellular TRIAC accumulation except for mmSLCO1B2 and drSlc10a1 (Supplementary Fig. S3D). Taken together, these results indicate that the mouse and zebrafish orthologs of the five human TRIAC transporters are capable of transporting TRIAC as well.

TRIAC accumulation in COS1 cells co-expressing CRYM and the mouse and zebrafish orthologs of SLC10A1, SLCO1B1, SLC22A6, and SLC22A8. 1 nM TRIAC accumulation experiments were performed in DPBS/0.1%glucose/0.1% BSA
Slc22a8 expression in postnatal mouse brain
Among TRIAC transporters identified, Slc22A8, which encodes the organic anion transporter 3 (Oat3), is of particular interest as it is expressed in the capillary endothelial cells in the mouse brain and transports multiple organic anions across mouse BBB. 32,33 We therefore studied the mRNA expression of Slc22A8 in the mouse CNS at different postnatal time points (P12, P21, and P120) in order to examine possible developmental changes in Slc22A8 expression. We also included Mct8/Oatp1c1 DKO mice in our study, as in these mice, the strongest thyromimetic action of TRIAC in the CNS was observed during the first three postnatal weeks. 6,7
Combined FISH analysis for Slc22a8 and the endothelial cell marker Pecam1 (CD31) in WT mice (Fig. 5) and DKO mice (Supplementary Fig. S6) at P12 and P21 revealed high Slc22a8 transcript levels in subsets of brain capillary endothelial cells (Fig. 5A and Supplementary Fig. S6A respectively), whereas in choroid plexus structures Slc22a8 specific FISH signals were only found in Pecam1 negative cells suggesting that choroid plexus epithelial cells express this transporter (Fig. 5B, Supplementary Fig. S6B). At postnatal day P21 in the choroid plexus and even more so at P120 in endothelial and epithelial cells, Slc22a8 signals were visibly decreased both in WT and DKO mice, indicating that Slc22a8 brain barrier expression declines in adulthood.
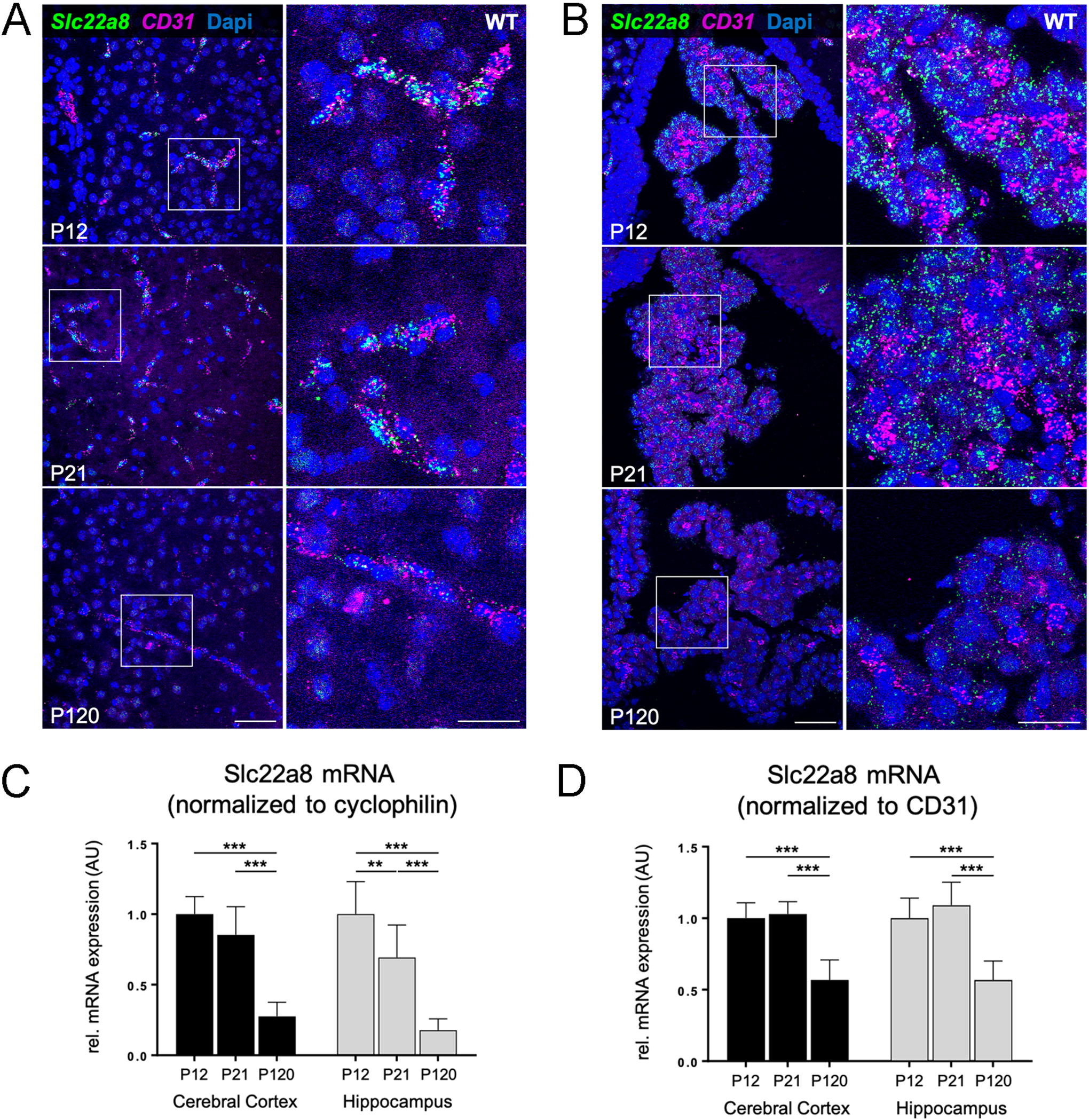
Temporal expression pattern of Slc22a8 mRNA in wild-type mouse brain barrier cells.
These findings were additionally confirmed by quantitative RT-PCR analysis of Slc22a8 expression in isolated cerebral cortices and hippocampi of WT mice at the same age points. Again, age-dependent decrease in Slc22a8 expression was observed in both brain areas when normalized to the house-keeping gene cyclophilin (Fig. 5C) and was even detected upon normalization to CD31 mRNA expression (Fig. 5D).
Discussion
In the present study, we identified SLC10A1, SLCO1B1, SLC22A6, SLC22A8, and SLC22A24 as well as their mouse and zebrafish orthologs as novel TRIAC transporters. In addition, we showed that the TRIAC transporter Slc22a8 is expressed in the capillary endothelial cells in the brain of Mct8/Oatp1c1 DKO mice, and this expression decreases in relation to postnatal age.
After its discovery in the 1950s by Pitt-Rivers, TRIAC has been administered to different patient populations, including patients suffering from myxedema coma and hypothyroidism, in which it could rapidly restore euthyroidism. 34 This was corroborated by findings in rodents, in which TRIAC was readily detected in multiple tissues such as adipose tissue, liver, heart, muscle, and kidney after administration. 35 Furthermore, TRIAC has been used as thyroid-stimulating hormone (TSH)-suppressive therapy in thyroid cancer, in resistance to thyroid hormone because of mutations in TRβ (RTHβ) and, recently, in MCT8 deficiency. 34,36 TRIAC may reach concentrations in the nM range in treated patients (unpublished observations). A common mechanism for TRIAC action in the abovementioned disorders entails a reduction in TSH, requiring uptake by the pituitary, and replacing thyroid hormone action in other tissues. Several transporters that increased intracellular TRIAC accumulation in the absence of BSA (i.e., SLC16A2, SLC38A1, SLC38A2, and ABCC5), but not in the presence of BSA, are expressed in the human pituitary gland. 37,38 This suggest that either the major pituitary TRIAC transporter is still unidentified or that multiple transporters, which at the individual level have a minor contribution, are jointly responsible for TRIAC entry into the pituitary.
Defective thyroid hormone transport across the BBB and/or BCSFB into neural cells likely underlies the neurodevelopmental delay in patients with MCT8 deficiency. 39 Different barriers, such as the brain endothelial cells whose tight junctions restrict paracellullar passage of compounds and the end feet of the astrocytes that line the basal side of these endothelial cells, should be overcome by TRIAC (or other thyroid hormone analogs) to be effective in the brain. SLC22A6 is expressed in astrocytes and oligodendrocyte precursor cells, whereas SLC22A8 is expressed in excitatory neurons and astrocytes, as indicated by single-cell RNA sequencing data. 37,38 However, they are not expressed in the brain endothelial cells. Therefore, although SLC22A6 and SLC22A8 have the potential to transport TRIAC into the neural cells, their exact roles in vivo await further clarification.
Of the five human TRIAC transporters identified, SLC10A1 and SLCO1B1 are liver-specific, whereas SLC22A6, SLC22A8, and SLC22A24 are predominantly expressed in kidney. 40 Rodent Slc22a6 and Slc22a8 are located at basolateral membrane of the kidney proximal tubule cells and contribute to xenobiotic and endogenous organic anion secretion. 41 –44 Their role of TRIAC clearance from the kidney, especially in the patients receiving TRIAC treatment, would be of interest for future studies.
All five human TRIAC transporters also transport T3, T4, or their sulfated forms. 18,45,46 None of the 40 transporters unknown to transport thyroid hormones were identified as robust T3 or T4 transporters. Only ABCG1 showed some T3 transport and ABCG5 some T4 transport, which was marginal compared with T3 and T4 transport by MCT8.
Studies in different animal models, including mice and zebrafish, suggest that TRIAC is able to enter the brain and, subsequently, exerts its thyromimetic function. 6,9 In zebrafish brain, Oatx is highly expressed, whereas Slc22a6l is expressed to a lesser extent. 31 Given the potent TRIAC transport by Oatx and Slc22a6l, they may explain the underlying route of TRIAC transport into brain enabling the rescue of hypomyelination in mct8-deficient zebrafish. 9 Neonatal initiation of TRIAC treatment in Mct8/Oatp1c1 DKO mice completely restored neurodevelopmental abnormalities, whereas the effectiveness strongly decreased when treatment started at P21. 8 Our observation that mRNA expression of the TRIAC transporter Slc22a8 decreases in an age-dependent manner may provide an explanation for this phenomenon. Given relevant differences in expression of TRIAC transporters across species, caution is needed in extrapolating the findings in mice to human beings. Moreover, it is difficult to predict physiological relevance based on in vitro studies. This would require genetic knockdown of transporters in animal studies, which could be challenging giving the potential redundancy of transporters that are able to facilitate TRIAC transport.
We acknowledge that our study has some limitations. First, we selected the candidate transporters for screening based on known substrates of these transporters and their expression at the BBB and BCSFB from transcriptome data. Our selection criteria may have missed relevant transporters. Second, some of the transporters in the selected panel lacked robust membrane expression in our model, precluding further testing. Future studies should explore, perhaps utilizing an unbiased transporter screening, whether other transporters are able to transport TRIAC. Third, we used binding proteins in our study, namely BSA in the incubation medium, to bind the substrates and CRYM intracellularly to accumulate substrates by preventing efflux. The affinity of TRIAC for BSA is unknown; however, half of TRIAC is distributed to albumin when added to human serum, indicating a relevant capacity for TRIAC to bind to albumin. 28 Yet differences between T3 and TRIAC in affinity for BSA would result in differences in the free concentration. We also do not know the affinity of TRIAC for CRYM. A lower affinity could result in a reduced accumulation of TRIAC compared with T3, independent from the transporter capacity. However, previous studies showed the accumulation of T4 and rT3 in the presence of CRYM, even though the affinity for CRYM is, respectively, 5- and 200-fold lower, 24,45,46 showing the robustness of the method. Nevertheless, the differences in binding affinities between T3 and TRIAC would complicate a direct comparison between the two substrates. However, the initial purpose of our study was to find transporters for TRIAC, not a comparison with T3 transport.
In summary, we identified five human TRIAC transporters with orthologs in zebrafish and mice, which are the first TRIAC transporters identified to date. These findings may contribute to further understanding the mechanism and potency of TRIAC in various disorders and in models thereof.
Footnotes
Authors’ Contributions
Conceptualization: Z.C., M.E.M., T.H., and W.E.V.; investigation: Z.C., S.Y., B.M., L.J.D.R., and S.L.; data analysis: Z.C. and B.M.; resources: W.E.V. and H.H.; writing—original draft: Z.C., M.E.M., W.E.V., B.M., and H.H.; writing—review and editing: all authors; supervision: M.E.M., W.E.V., R.P.P., and H.H.; funding acquisition: R.P.P. and H.H.
Author Disclosure Statement
Erasmus Medical Center receives royalties from Egetis Therapeutics on the commercialization of TRIAC. None of the authors have personal benefits from any royalties.
Funding Information
This study is funded by a Vidi grant (
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5