
Abstract
Background:
Papillary thyroid cancer (PTC) is the predominant subtype of thyroid cancer (
Methods:
Our previous investigation of 17 AD PTC families led us to conduct a deeper analysis on one family (Family Q) with whole-genome sequencing data from 3 PTC-affected individuals. In addition, 323 sporadic
Results:
We found truncating PDPR splice donor variants (NM_017990.4:c.361 + 1G>C) in all affected PTC Family Q members, and another PDPR splice donor variant (NM_017990.4:c.443 + 1G>C) in a sporadic PTC case. In addition, an ultra-rare missense variant was found in an FAP-PTC patient. The PDPR-deficient cells presented with elevated phosphorylation of pyruvate dehydrogenase and altered glucose metabolism, implying that PDPR plays an essential part in regulating glucose metabolism in thyroid cells.
Conclusions:
Our finding of novel truncating germline variants in PDPR in Family Q and additional cohorts suggests a role for PDPR loss in PTC predisposition. Also, somatic and RNA sequencing from the thyroid carcinoma (Firehouse Legacy) data showed that PDPR gene expression is much lower in
Introduction
Thyroid cancer (THCA) is the most common endocrine malignancy and is the 12th most common cancer in the United States. 1 More than 90% of all THCAs are derived from the thyroid follicles and are collectively referred to as non-medullary thyroid carcinomas (NMTC). Papillary thyroid cancer (PTC) is the most common form of NMTC. 2 While most NMTC are sporadic, familial forms are estimated to account for 3%–15%. 3,4 Familial NMTC is suspected in families with at least three close relatives, usually in multiple generations and with an absence of syndromic features. Inheritance appears to be dominant with incomplete penetrance. 4 Familial forms may represent more aggressive disease. 5,6
Hereditary tumor predisposition syndromes only account for a small percentage (∼5%) of familial NMTC. For example, individuals with familial adenomatous polyposis (FAP) have been noted to have an increased risk for
The genetic etiology of nonsyndromic familial NMTC has been elusive. We previously conducted a study on 17 THCA families to search for germline variants predisposing to PTC. 8 Our present investigation focuses on the pyruvate dehydrogenase phosphatase regulatory subunit (PDPR) gene identified as one of the candidate genes segregating in Family Q (Fig. 1 and Supplementary Table S1). We report genetic data on PDPR in a cohort of sporadic THCA cases (Avatar, n = 323) and 12 syndromic FAP-PTC cases. We identify a truncating PDPR gene variant segregating with familial PTC, and other PDPR variants in independent cohorts. Our functional experiments suggest that PDPR loss leads to altered glucose metabolism, resembling previous observations from different cancer types. 9 –11

Pedigree of Family Q. Arrow indicates the index person. Stars denote family members included in whole-genome sequencing. Individuals II.7, III.8, III.10, IV.5, and IV.8 are heterozygous of the PDPR c.361 + 1G>C variant (plus sign), whereas II.8, IV.2, IV.7, and IV.9 are homozygous for the reference allele (minus sign). The individual IV.6, affected with parotid cancer at 14 years of age, is likewise a heterozygous of the PDPR c.361 + 1G>C variant. A diamond shape structure shows the number of the healthy children. Family Q shows 11 different cancer types, including very rare types of salivary gland cancer and throat cancer (no samples available), and parotid cancers. Please see Supplementary Table S1 for clinical characteristics of the individual family members.
Materials and Methods
Patient cohorts
The Ohio State University (OSU) Institutional Review Board approved this study, and all the participating subjects gave their written informed consent. Family Q was consented to OSU protocol 1998C0143, and Avatar cases were consented to OSU protocol 2013H0197. In a cohort of 17 PTC families, whole-genome sequencing (WGS) on blood DNA was performed on 3 PTC-affected members from one family (Family Q),
8
with the purpose to find out which of the gene variant(s) was predisposing to PTC. We also accessed whole-exome sequencing (WES) data on germline DNA from 323 sporadic THCA cases (Avatar cohort), with some cases also having RNA-seq data available for analysis (Avatar A). In addition, germline and matching
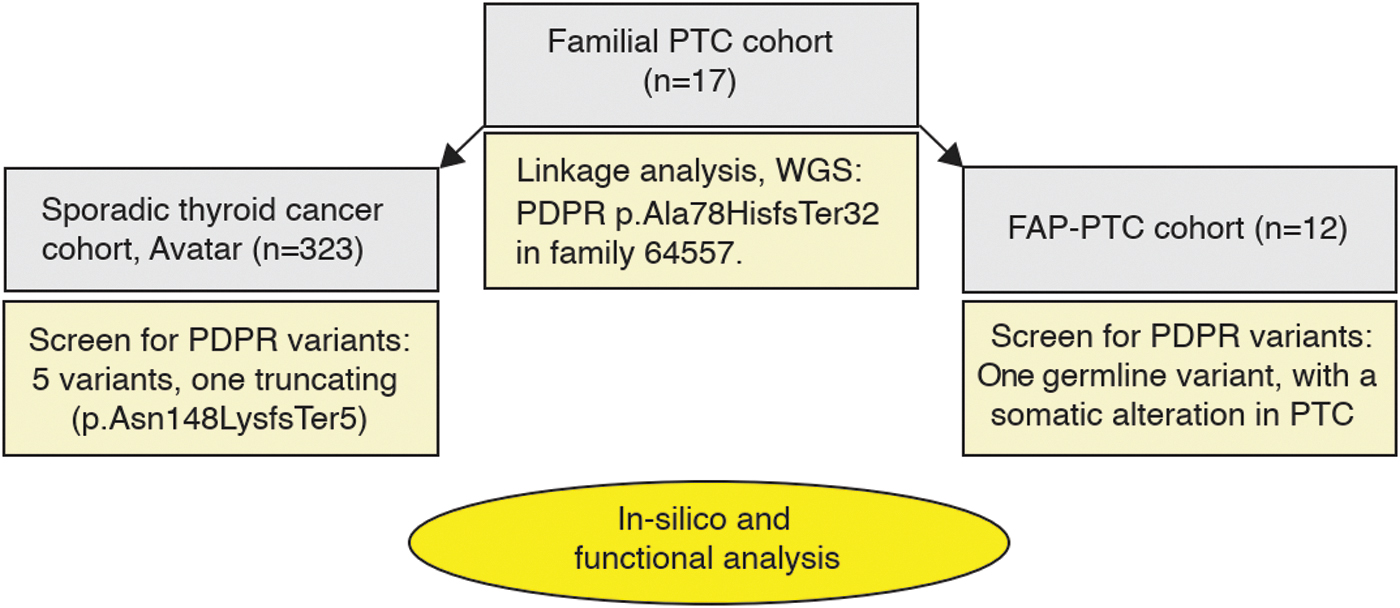
A schematic representation of the workflow of this investigation. Gray boxes indicate the investigated cohorts, while yellow boxes and the oval detail the methodology and identified PDPR variants.
PDPR Variants of a Family Q, an Avatar, and a Familial Adenomatous Polyposis–Papillary Thyroid Cancer Cohort
ACMG, American College of Medical Genetics and Genomics; FAP, familial adenomatous polyposis; G, germline; N/A, not available; PTC, papillary thyroid cancer; S, somatic; VUS, variant of unknown significance.
Sanger sequencing, reverse transcription–polymerase chain reaction, and TOPO TA cloning
Genomic DNA was Sanger sequenced from Family Q members and from the individual with sporadic THCA to confirm the presence of PDPR variants. We sequenced all available PTC-affected as well as non-PTC-affected samples to identify variants segregating in Family Q. The effect of the identified PDPR variants on splicing was assessed by reverse transcription (RT)–polymerase chain reaction (PCR) (see the Supplementary Data and Supplementary Fig. S2).
Cell culture and CRISPR-Cas9 targeting of PDPR in N-Thyori3-1 and TPC1 cells
Cell culture and CRISPR-Cas9 methods are described in the Supplementary Data.
Seahorse assay and Tracer liquid chromatography mass spectrometry flux metabolomics
Western blotting, quantitative RT-PCR, the tracer liquid chromatography mass spectrometry flux metabolomics and statistical analysis, and the Seahorse assay are described in the Supplementary Data.
RNA expression analysis of THCA cohorts and sporadic sample (Avatar A)
RNA expression analyses are described in the Supplementary Data.
In silico analysis (SWISS modeling)
We applied in silico modeling analysis 13 to predict the effects of PDPR germline variants on PDPR protein compared with the wild-type (wt) protein. Detailed description is available in the Supplementary Data.
Results
Identification of PDPR as the primary candidate gene in Family Q
PTC families investigated in our previous work typically revealed multiple chromosomal regions of tentative linkage. Family Q constituted an exception in that a single region located on chromosome 16 was pinpointed and five variants in four genes (AARS, ITGAD, CHD11, and PDPR) from that region fulfilled our stringent selection criteria. 8 Many different cancer types are represented in this family (Fig. 1. and Supplementary Table S1), although thyroid and parotid cancers were overrepresented. We expanded the previous study by sequencing samples from additional affected members (e.g., a parotid gland cancer patient) as well as healthy family members (Fig. 1). This analysis allowed us to exclude AARS, ITGAD, and CHD11 from the candidate gene list (Supplementary Table S2). PDPR c.361 + 1G>C is the only variant that cosegregated with disease in all four studied PTC-affected members as well as in a parotid gland cancer patient (IV.6) and was absent in all three healthy family members analyzed (Fig. 1). Unfortunately, no tumor DNA samples from THCA or salivary gland cancer patients were available for analysis (Supplementary Table S1). The workflow of the present study is summarized in Figure 2.
Occurrence of PDPR germline variants in a cohort of sporadic PTC cases (“Avatar”)
We screened a cohort of 323 sporadic PTCs by WES for germline variants in PDPR. In total, the cohort harbored 5 PDPR single nucleotide variants in 5 samples including a canonical splice site variant, PDPR c.443 + 1G>C, in a patient sample referred to herein as Avatar A. We used VarSome 14 to predict the consequences of the identified variants in silico by using three-tier annotations. The predominant predictions were variants of unknown significance (VUS) or pathogenic, given by VarSome's American College of Medical Genetics and Genomics (ACMG) classification. The PDPR c.443 + 1G>C splice site variant was predicted likely pathogenic (Table 1).
Experimental and in silico validation of the PDPR variants
Sanger sequencing confirmed the PDPR c.361 + 1G>C variant in Family Q and the PDPR c.443 + 1G>C variant in the Avatar A sample, which are represented in Figure 3A. As evident from the population frequencies (Table 1), the PDPR c.361 + 1G>C variant is absent in the average population, whereas PDPR c.443 + 1G>C is ultra-rare.
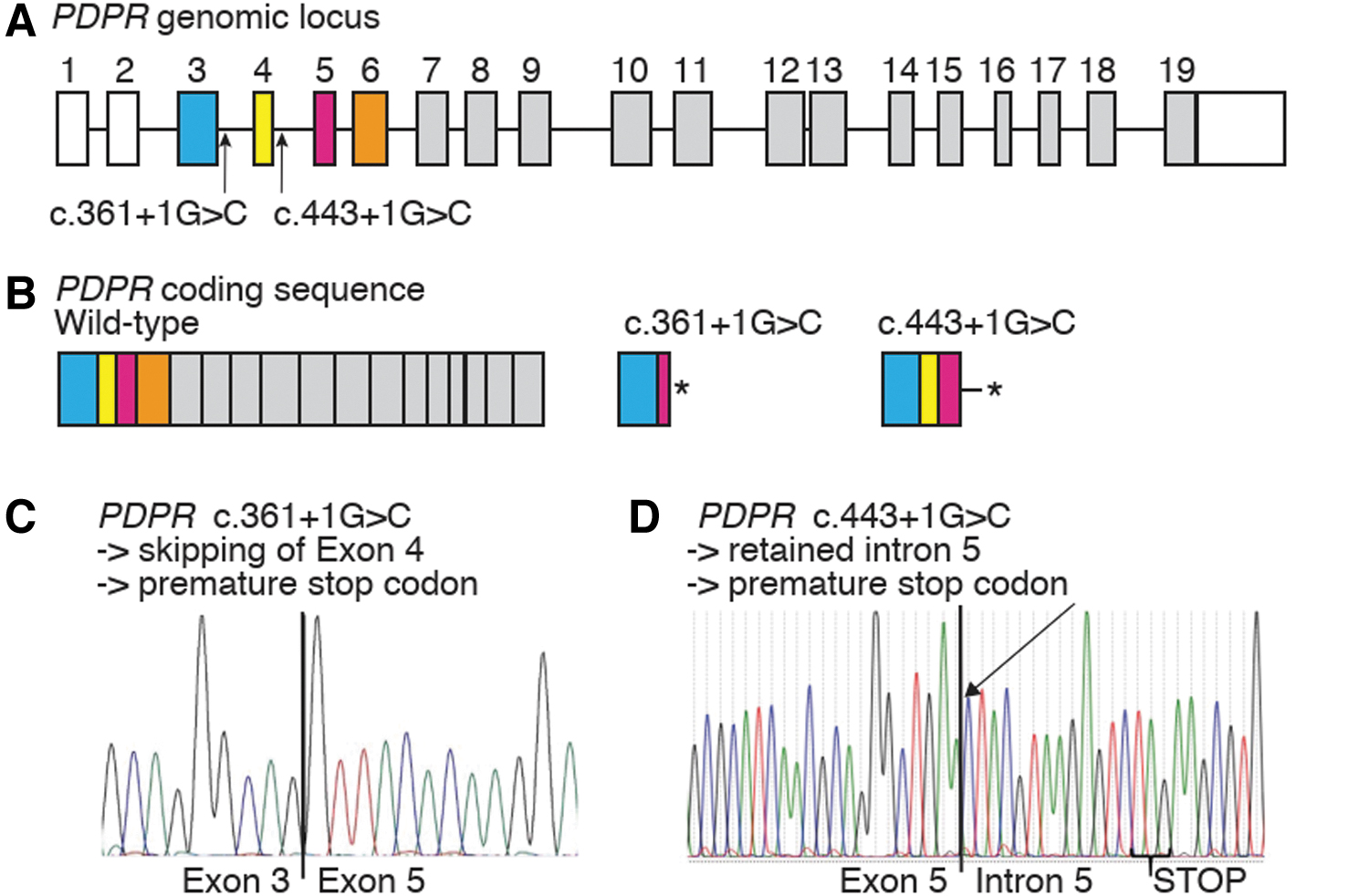
Sequencing analysis of PDPR mutations. (
PDPR c.361 + 1G>C in Family Q was predicted to result in altered splicing (Supplementary Fig. S1A). To verify the predicted effects, we performed RT-PCR analysis on blood RNA samples from 3 PTC patients (lanes 2, 3, and 4), 3 healthy members (lanes 5, 6, and 7) from Family Q, and a nonfamily member (lane 8) (Supplementary Fig. S1B). All healthy samples produced a >300 bp band corresponding to the predicted fragment including exon 4. All PTC patients presented with an additional shorter product corresponding to the loss of the 134 bp exon 4.
The skipping of exon 4 (coding exon 2) was confirmed by Sanger sequencing of the cDNA cloning product from a PDPR c.361 + 1G>C variant carrier (Fig. 3C). The aberrant splicing is predicted to lead to the incorporation of 32 aberrant amino acids before a downstream premature stop codon, likely resulting in a truncated PDPR protein (p.Ala78HisfsTer32*) (Fig. 3B). Upon Sanger sequencing of the TOPO TA colonies from the c.361 + 1G>C variant carrier, 17 clones with the wt sequence and 10 clones with the mutant sequence were identified. This suggests a degree of underrepresentation of the mutant alleles in cDNA (compatible with possible nonsense-mediated RNA decay), although statistically nonsignificant.
Sanger sequencing of the cDNA cloning product of a PDPR c.443 + 1G>C variant carrier from the Avatar cohort showed a retention of intron 5 (Fig. 3D), which is predicted to lead to truncated PDPR protein (p.Asn148LysfsTer5*) (Fig. 3B). The entire intron 5 was present in the cDNA Sanger sequence of the Avatar A sample, depicted schematically in Supplementary Figure S1C. Ten colonies from the TOPO TA cloning assay for Avatar A were picked for Sanger sequencing. Three of them represented the mutant allele, while seven contained the wt allele.
For the Avatar A sample, we had the additional opportunity to visualize the PDPR transcripts reads between exons 4 and 7 (coding exons 2–5) in RNA sequencing data. The integrative genomics viewer and its Sashimi plot tool revealed no reads between coding exons 5 and 6 because the entire 93 bp intron 5 was present in the transcript with no splicing junction occurring between exons 5 and 6, thus contrasting with a healthy control individual (Supplementary Fig. S1D).
SWISS modeling of the wt PDPR amino acid sequence and the predicted mutant PDPR amino acid sequence of Family Q and the Avatar A sporadic sample verifies the abnormal three-dimensional shape of the mutant PDPRs (Supplementary Fig. S2B, C, respectively), also lacking most of the amino acids of the wt PDPR.
PDPR variant screen in a cohort of syndromic PTC cases
Recently, we performed a study to clarify why FAP patients are prone to THCA. 12 By using WGS of normal thyroid tissue or blood and matching THCA, we investigated 12 FAP patients from the United States and Finland (all confirmed APC pathogenic variant carriers) who also had THCA. One sample out of 12 (8.3%) had a heterozygous rare PDPR c.1372C>T, p.Arg458Cys variant. 12 Our present SWISS modeling revealed that this PDPR variant was likely to result in a structurally abnormal protein compared with the wt protein (Supplementary Fig. S2D). Moreover, we detected an additional ultra-rare somatic PDPR variant (c.1373G>A, p.Arg458His) in the thyroid tumor of the patient (Table 1). The somatic PDPR variant (p. Arg458His) in the FAP-PTC case may represent an acquired second hit contributing to a multistep model of tumorigenesis, 15 especially considering that this patient's thyroid tumor was lacking a somatic APC pathogenic variant (and based on the in silico prediction from the SWISS model). Most of the other FAP patients had somatic APC pathogenic variants in the corresponding THCA. 12 When applying the ACMG variant classification criteria, this somatic PDPR alteration is currently a VUS (Table 1).
Analyses of The Cancer Genome Atlas Program and OSU cohorts for PDPR RNA expression and somatic variants
Poorly differentiated and anaplastic thyroid cancers 16 and thyroid carcinoma (The Cancer Genome Atlas Program [TCGA], Firehouse Legacy) somatic cohorts (n = 625 samples) had somatic PDPR variants in two cases of thyroid malignancies (0.3%). These included a homozygous deletion in one case and a missense-type variant, p.His688Leu, in the other case. 17
To evaluate the expression levels of PDPR in tumors compared with matching normal tissue, we evaluated RNA sequencing data for a cohort of 59 TCGA (Firehouse Legacy) THCA with normal tissue pairs. Interestingly, the tumor group had significantly (p < 0.0001) lower PDPR gene expression than normal thyroid samples (Supplementary Fig. S3). These results were replicated in our OSU cohorts with RNA sequencing data from 16 sporadic THCA tumor and adjacent normal pairs. 18 This cohort also showed significantly lower expression of PDPR in matching tumor versus adjacent normal thyroid tissue (Supplementary Fig. S3). The results indicate that decreased PDPR expression is a common phenomenon in THCA compared with normal thyroid tissue and suggest reduced PDPR levels as a possible contributing factor in thyroid tumorigenesis.
Functional analysis of PDPR loss in cell lines
Increased inhibitory phosphorylation of pyruvate dehydrogenase (PDH) complex is frequently observed in cancer cells and associated with reduced mitochondrial metabolism and increased aerobic glycolysis. 19 PDPR is the regulatory subunit of pyruvate dehydrogenase phosphatase (PDP) complex, which dephosphorylates and activates PDH-Ei alpha, the regulatory subunit of the PDH complex. 20 Currently, the specific mechanism by which PDPR loss impacts PDH activity and glucose metabolism in thyroid cells is unknown. To address this, we generated PDPR-deficient pools of the nontransformed thyroid cell line Nthy-ori 3-1 21,22 as well as a THCA cell line (TPC1) using the CRISPR-Cas9 system with two independent gRNAs targeting PDPR. The success of the targeting was validated using Sanger sequencing (Supplementary Fig. S4A, B) and quantitative PCR (Supplementary Fig. S4C). Interestingly, both cell lines exhibited increased phosphorylation of PDH-E1 alpha upon PDPR loss, likely via reduced activity of PDP (Fig. 4A, B).
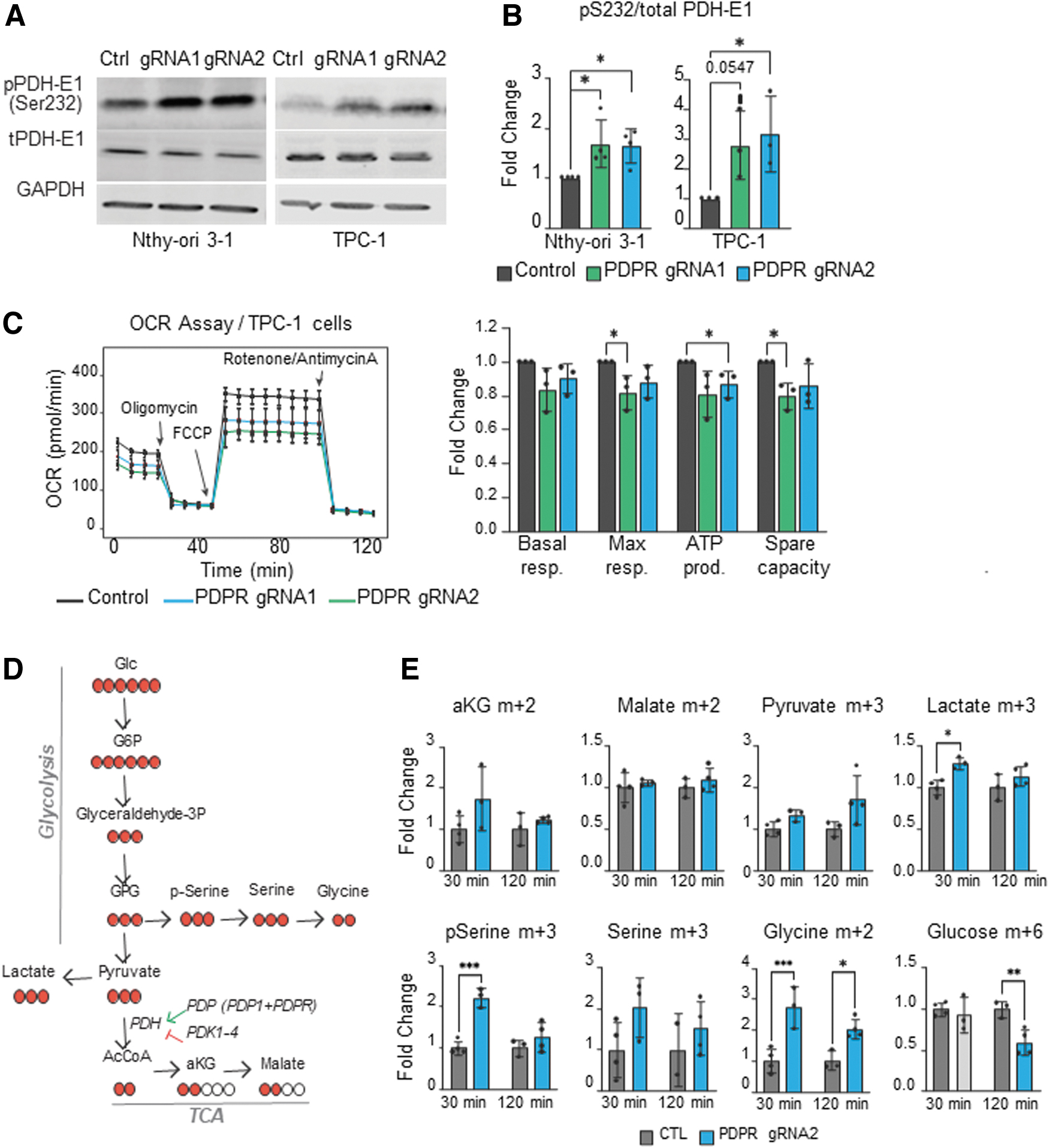
Increased PDH-E1 phosphorylation as a result of PDPR deficiency. (
PDH complex catalyzes the conversion of glycolytic pyruvate to acetyl coenzyme A (acetyl-CoA), a crucial metabolite used by the tricarboxylic acid (TCA) cycle. To address whether the increased PDH-E1 alpha phosphorylation reflects mitochondrial activity and thus the balance of oxidative to glycolytic metabolism in PDPR-deficient cells, we measured the oxygen consumption rate (OCR) using the Seahorse assay. Indeed, while basal respiration was not significantly altered, PDPR loss reduced the maximal respiratory capacity, adenosine triphosphate production, and spare respiratory capacity in TPC1 cells (Fig. 4C). However, in the nontransformed Nthy-ori 3-1 cells, PDPR loss did not induce significant changes in the OCR (Supplementary Fig. S5). Thus, to investigate the metabolic changes reflecting the loss of PDPR and increase of PDH phosphorylation in Nthy-ori 3-1 cells, we performed glucose tracing using 13 C labeling (Fig. 4D).
Consistently with the OCR assay, carbon flux into the TCA cycle via PDH complex (aKG m + 2 and malate m + 2) was not significantly changed; however, the labeled lactate m + 3 levels were increased at 30 minutes after 13 C-Glucose pulse, suggesting a shift toward glycolytic metabolism (Fig. 4E). In addition, the amount of labeled metabolites in the serine–glycine biosynthesis pathway (pSerine m + 3 and Glycine m + 2), a glycolytic shunt pathway frequently activated in cancer, 23 was increased compared with the control cells (Fig. 4E). These results indicate that the loss of PDPR in thyroid cells is associated with increased PDH phosphorylation, resulting in reduced oxidative metabolism and a shunt of the glucose-derived carbons into serine–glycine metabolism. These metabolic changes could be functionally involved in predisposition to THCA (Fig. 5).

Schematic representation of the findings.
Discussion
We herein describe two different truncating PDPR variants in a PTC family and a sporadic PTC case. In addition, we find an ultra-rare somatic PDPR variant in a syndromic form of PTC case (FAP-PTC). Our metabolic and functional data show dysregulation of glucose metabolism, outlining a possible mechanism of pathogenicity for PDPR loss-of-function variants predisposing for a subset of PTC.
The mechanisms by which germline events predispose to PTC are weekly characterized. Despite extensive efforts, only a few proven or probable predisposing variants have so far been reported, which is why we undertook this investigation. In our recent WGS study of 17 families displaying PTC in at least 3 close relatives, stringent filtering criteria were applied in search for predisposing gene variants. Family Q is the focus of the present study. We found a PDPR p. Ala78HisfsTer32 segregating perfectly with the PTC, and it was also present at a young age at onset patient with parotid cancer. In the sporadic setting (Avatar), we identified five rare or ultra-rare variants in PDPR gene. One of those variants (c.443 + 1G>C, Table 1) resulted in a splicing defect leading to a truncated mRNA, suggesting loss of function, such as Family Q.
We propose that in Family Q, PDPR is implicated in PTC predisposition because the c.361 + 1G>C variant segregates with PTC and with parotid cancer and was absent in unaffected/healthy individuals. This splice donor variant of Family Q leads to skipping of exon 4 (coding exon 2) of the PDPR gene, creating a premature stop codon (Fig. 3B). The splice donor variant c.443 + 1G>C in one of the sporadic PTC patients (Avatar A) leads to retention of intron 5 and premature stop codon as well (Fig. 3B). Both aforementioned variants seem to result in protein truncation and highly aberrant three-dimensional configuration by SWISS modeling (Supplementary Fig. S2). These findings suggest that constitutional PDPR variants explain a rare subset of familial and sporadic PTC. Moreover, in syndromic FAP PTC, PDPR variants may contribute to the multistep process of thyroid tumorigenesis, as suggested by our finding of a germline and somatic PDPR variant in one of the FAP-PTC cases (Table 1).
Our somatic findings from the TCGA and RNA sequencing data might indicate that PDPR acts like a typical tumor suppressor gene (Supplementary Fig. S3). PDPR gene expression is much lower in tumor tissue compared with matching normal tissue in the TCGA cohort (n = 59) and in 16 sporadic tumor and adjacent normal pairs.
Altered metabolism is a hallmark of cancer, typically involving glucose metabolism shifting toward glycolytic pathways. 24 For example, conversion of the glycolytic end product, pyruvate, into acetyl-CoA by the mitochondrial pyruvate dehydrogenase complex (PDC) is frequently reduced in cancer cells via increased inactivating phosphorylation of the PDH E1 subunit by pyruvate dehydrogenase kinases 1–4. 25 PDC is activated by PDP complex consisting of the catalytic subunit PDP1 and the regulatory subunit PDPR. While PDPR has been shown to decrease the sensitivity of PDP1 to Mg2+ ions, 26 the role of PDPR in glucose metabolism and carcinogenesis is not well understood.
THCA has been reported to manifest metabolic changes including alterations in glucose metabolism. 27 In addition, expression of proteins regulating serine–glycine metabolism differs among THCA subtypes. 28 Notably, the serine–glycine pathway is a crucial source of nucleotides necessary for cancer cells including THCA. 27 It has been recently reported that anaplastic THCA cells upregulate the one-carbon metabolism, and inhibition of this pathway could be a potential therapeutic target. 9 However, it is not clear if alterations in metabolic genes can predispose to PTC.
In this study, we targeted PDPR using CRISPR-Cas9 in normal thyroid and THCA cells and examined its consequences on glucose metabolism. We observed increased phosphorylation of PDC upon PDPR loss, suggesting that PDPR is required for the normal dephosphorylation activity of PDP. This result is consistent with the notion that many cancer types including PTC show alterations in glucose metabolism. Interestingly, while the TPC1 cells did show reduced oxidative metabolism, in the nontumorigenic Nthy-Ori 1 cells, PDPR loss led to increased shift of glucose-derived carbons into the serine–glycine pathway. Our results suggest that PDPR deficiency-induced metabolic alterations may be mechanistically involved in a subset of familial occurring PTCs.
Based on evidence described above, we suggest that variants in the PDPR gene leading to reduced protein expression or function could be major determinants of PTC occurrence in Family Q, the Avatar A case, and the FAP-PTC case. However, we cannot rule out the possibility that other gene variants, such as AARS, ITGAD, and CHD11 (Supplementary Table S2), might have a separate minor or together combinational role in PTC occurrence in the Family Q. The Avatar cohort was explored by WES targeting protein coding variants only, overlooking possible non-coding variants and large genomic rearrangements, which might in part account for the rarity of constitutional PDPR variants in that cohort. Our observation of low somatic variant rates but frequently reduced mRNA expression in PTC tumors from TCGA (Supplementary Fig. S3) might imply a role for non-coding or post-transcriptional regulatory mechanisms in PDPR-associated tumorigenesis.
Footnotes
Acknowledgments
We wish to acknowledge Prof. Albert de la Chapelle (1933–2020) and Dr. Huiling He for their contributions to the origins of this project and for their support and expertise. The facilities and expertise of FIMM Metabolomics, supported by HiLIFE and Biocenter Finland, are gratefully acknowledged.
Data Availability
This study includes no data deposited in external repositories. The familial patient consents do not allow sharing of raw sequencing data. All variants can be found in the article and Supplementary Data. The Avatar data used in this research were generated through private funding by Aster Insights (www.asterinsights.com) in collaboration with the Oncology Research Information Exchange Network (ORIEN, www.oriencancer.org). Requests for access to the data used in this study can be submitted here at ![]()
Authors' Contributions
M.D.R., P.P., T.T.N., and S.O. designed the experiments and supervised the project. D.F.C., M.S., T.T.N., S.S., A.I.N., and S.O. performed the molecular laboratory experiments, in silico analysis, and the results interpretation. A.S.Y., S.L., and T.T.N., performed genome sequencing data analysis and the results interpretation. P.B. contributed NMTC Family Q and sporadic samples clinical information. M.D.R. and P.P. were responsible for acquisition of the funding for the project. M.S., P.B., M.D.R., P.P., S.O., S.L., and T.T.N. wrote the original article draft.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the National Cancer Institute Grants P50CA168505 (M.D.R.) and P30CA16058 and Jalmari and Rauha Ahokas Foundation (T.T.N.) and the Jane and Aatos Erkko Foundation, the Academy of Finland (no. 330606), Cancer Foundation Finland sr, and the Sigrid Juselius Foundation (P.P.).
Supplementary Material
Supplementary Data
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5