
Abstract
Background:
Congenital hypothyroidism (CH) is the most common neonatal metabolic disorder. In patients with CH in China, thyroid dyshormonogenesis is more common than thyroid dysgenesis; however, the genetic causes of CH due to thyroid dyshormonogenesis remain largely unknown. Therefore, we aimed at identifying novel candidate causative genes for CH.
Methods:
To identify novel CH candidate genes, a total of 599 patients with CH were enrolled and next-generation sequencing was performed. The functions of the identified variants were confirmed using HEK293T and FTC-133 cell lines in vitro and in a mouse model organism in vivo.
Results:
Three pathogenic contactin 6 (CNTN6) variants were identified in two patients with CH. Pedigree analysis showed that CH caused by CNTN6 variants was inherited in an autosomal recessive pattern. The CNTN6 gene was highly expressed in the thyroid in humans and mice. Cntn6 knockout mice presented with thyroid dyshormonogenesis and CH due to the decreased expression of crucial genes for thyroid hormone biosynthesis (Slc5a5, Tpo, and Duox2). All three CNTN6 variants resulted in the blocking of the release of the Notch intracellular domain, which could not translocate into the nucleus, impaired NOTCH1 transcriptional activity, and decreased expression of SLC5A5, TPO, and DUOX2. Further, we found that DTX1 was required for CNTN6 to promote thyroid hormone biosynthesis through Notch signaling.
Conclusions:
This study demonstrated that CNTN6 is a novel causative gene for CH through the mediation of thyroid hormone biosynthesis via Notch signaling, which provides new insights into the genetic background and mechanisms involved in CH and thyroid dyshormonogenesis.
Introduction
Congenital hypothyroidism (CH) is the most common congenital endocrine disorder, affecting 1 in 2000–4000 newborns worldwide 1 –4 In 2021, a large-scale epidemiological study showed a high prevalence of CH in China (1 in 2144). 5 Without prompt treatment, CH can lead to severe and irreversible growth retardation and neurological deficits. Based on etiology, CH can be divided into two groups: thyroid dysgenesis and thyroid dyshormonogenesis. Thyroid dysgenesis accounts for 85% of Caucasian cases and is mainly caused by genetic variants in genes, including NKX2-1, FOXE1, PAX8, NKX2-5, TSHR, TBX1, CDCA8, HOXD3, and HOXB3, while 15% of cases are due to thyroid dyshormonogenesis, which is associated with SLC5A5/NIS, SLC26A4/PDS, SLC26A7, TPO, DUOX1, DUOX2, DUOXA2, IYD/DEHAL1, TG, and TSHR. 6
Recent studies have revealed that thyroid dyshormonogenesis is the primary cause of CH in China; however, variants of these known genes account for only 50% of patients. 7 –10 Therefore, the pathogenesis of CH is largely unknown. There is an urgent need to identify novel CH pathogenic genes and clarify their role in the thyroid gland.
Recently, the Notch signaling pathway has been implicated in thyroid cell fate determination and thyroid-specific gene expression. 11 –13 Notch signaling is dependent on a ligand triggering receptor proteolysis and release of the Notch intracellular domain (NICD), an active Notch fragment, into the cytoplasm. The NICD translocates to the nucleus and associates with a DNA binding protein CSL (CBF1, Suppressor of Hairless, Lag-1) to assemble a transcription complex that activates downstream target genes, including HES1. 14
Classical Notch receptor ligands include Delta, Serrate/Jagged, and Lag2 (DSL). Contactin 6 (CNTN6) has been identified as a non-canonical ligand of NOTCH1, activating non-canonical Notch signaling. 15
CNTN6 is the sixth member of the contactin family of immunoglobulin cell adhesion proteins, which exhibit a high degree of amino acid overlap (40–60%), and each contain six immunoglobulin-like domains and four fibronectin type III-like domains that are linked to the plasma membrane through a glycosylphosphatidylinositol-anchor. 16 There has been limited research on CNTN6; it is suggested that it may be involved in brain development.
Cntn6-deficient mice show delayed corticospinal tract development, misoriented apical dendrites in layer V of the visual cortex, and increased neuronal cell death during development. 17 –20 CNTN6 abnormalities are closely associated with the occurrence of autism spectrum disorders and intellectual disabilities. 21,22 As the non-canonical ligand for NOTCH1, CNTN6 triggers nuclear translocation of NICD and promotes oligodendrogliogenesis from progenitor cells and differentiation of oligodendrocyte precursor cells via Deltex1 (DTX1). 15
The Genotype-Tissue Expression (GTEx) dataset in the Human Protein Atlas (HPA) database shows that CNTN6 exhibits high expression and tissue specificity in the thyroid and brain. However, no studies have explored the role of CNTN6 in the thyroid.
In this study, we identified a novel candidate causative gene, CNTN6, in two families with CH. Through in vitro and in vivo experiments, we confirmed that CNTN6 affected thyroid hormone biosynthesis through the Notch signaling pathway with the involvement of DTX1. Three CNTN6 variants derived from patients with CH were found to damage the release and nuclear translocation of NICD (an active Notch fragment), leading to the downregulation of key genes (SLC5A5, TPO, and DUOX2) involved in thyroid hormone biosynthesis, ultimately resulting in CH.
Methods
Clinical subjects
In total, 599 patients with CH were recruited for next-generation sequencing and through collaboration with multiple hospitals in China. All subjects provided informed consent using protocols approved by the Ethics Committee of Shanghai Ninth People's Hospital affiliated with Shanghai Jiao Tong University School of Medicine (approval number 2016-76-T33). Diagnosis of CH was based on the same criteria according to our previous studies 7 (details in Supplementary Methods and Supplementary Fig. S1).
Generation of Cntn6-knockout mice
All animal studies were approved by the Ethics Committee of Shanghai Ninth People's Hospital affiliated with Shanghai Jiao Tong University School of Medicine (approval number SH9H-2021-A99-1). The Cntn6 knockout (KO) mice of the C57BL/6N strain were generated by Cyagen Biology. Briefly, two guide RNAs (gRNAs) were designed in intron 3 (GGAAGGTCTCTATCCTTTATAGG) and intron 4 (TGACTAAAGTTCTATTATCCAGG) to KO Cntn6 exon 4. Subsequently, Cas9 was combined with sgRNAs that targeted introns 3 and 4 and injected into the oviduct at E0.7 in plug-confirmed mice.
At the age of five days, the mice were genotyped using DNA extracted from the clipped toes. The F0 generation chimera founder was then mated with wild-type (WT) mice to produce F1 heterozygous (HET) mice. Finally, F1 generation HET male and female mice were crossed to obtain F2 generation mice containing a homozygous (HOM) mutant.
Statistical analysis
Statistical analyses were carried out with IBM SPSS Statistics 25. Quantitative data were reported as mean ± standard error of the mean. Depending on the situation, intergroup comparisons were carried out using the Student's unpaired t-test for normally distributed data or the Mann–Whitney U test for non-normally distributed data. Categorical data were presented as percentages and were compared between groups using the chi-square test or Fisher exact test as appropriate. A p-value of <0.05 was considered statistically significant.
Additional methods are presented in Supplementary Materials.
Results
Identification of CNTN6 variants in two patients with CH
We recruited 599 patients with CH: 311 males and 288 females. Detailed clinical characteristics are shown in Supplementary Table S1. The mean values of serum free triiodothyronine (fT3), free thyroxine (fT4), and thyrotropin (TSH) levels at diagnosis were 3.09 pg/mL, 0.73 ng/dL, and 78.37 μIU/mL, respectively. Thyroid morphology was evaluated in 299 patients with CH, and most of the patients (83%) had hypothyroidism with gland-in situ (GIS) (Supplementary Table S1).
Three hundred eight patients with CH (51.4%) carried none of the pathogenetic variants in the 21 known CH genes identified in our previous study. 7 In these patients, we conducted a series of rigorous quality control and bioinformatics analyses. Combined with the genetic pattern of CH, we performed familial genetic analysis, and three pathogenic CNTN6 variants were identified in two patients from two CH pedigrees (Fig. 1A, B).
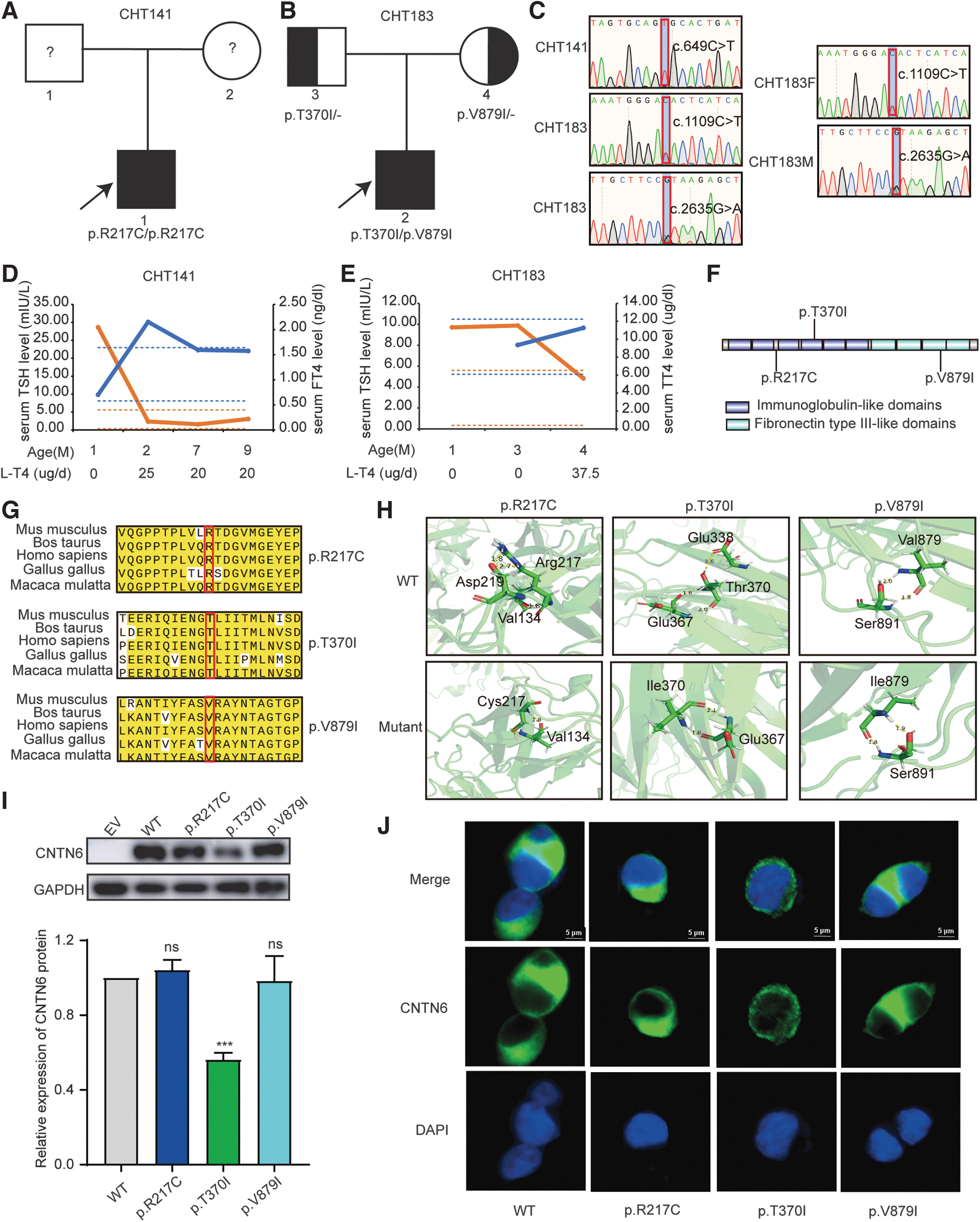
Identification of CNTN6 variants in CH patients. (
Two probands' thyroid ultrasound results showed GIS and normal volume (Table 1). We conducted nine- and four-month clinical follow-ups for proband CHT141 and proband CHT183, respectively, whose intelligence was assessed by the Developmental Screening Test. Detailed clinical information and thyroid function follow-up of the two probands are shown in Table 1 and Figure 1D, E, respectively. Following timely diagnosis and treatment, both patients currently show no apparent signs of intellectual or developmental delays.
Detailed Clinical Information of the Two Congenital Hypothyroidism Patients with CNTN6 Biallelic Variants
TSH, thyrotropin.
Further, the transmission patterns of CNTN6 in the two pedigrees were investigated by Sanger sequencing. We observed that proband CHT141 carried homozygous CNTN6 variants (p.R217C), and proband CHT183 carried compound HET CNTN6 variants (p.T370I/p.V879I). All variants of patients were inherited from their parents who are euthyroid with a normal-sized thyroid gland, which was in line with an autosomal recessive inheritance manner (Fig. 1A–C).
The three CNTN6 variants from two probands were highly conserved among species (Fig. 1G). The p.T370I variant was located at the fourth immunoglobulin-like domain, and the p.V879I variant was located at the third fibronectin type III-like domain (Fig. 1F). Further, we predicted the three-dimensional structure of the WT and three mutant CNTN6 proteins in silico tools. The p.R217C variant disrupted the hydrogen bond interaction between arginine 217 and aspartic acid 219. The p.T370I variant resulted in the loss of the hydrogen bond link between threonine 370 and glutamic acid 338 (Fig. 1H). By transfecting WT and mutant CNTN6 plasmids into HEK293T cells, we found that the T370I variant significantly reduced the protein expression level of CNTN6, while the expression level of p.R217C and p.V879I mutant proteins was akin to WT (Fig. 1I). The immunofluorescence results indicated that all three mutant and the WT proteins exhibited membrane localization tendencies (Fig. 1J). Specific information regarding these three variants is listed in Supplementary Table S2.
Cntn6-deficient mice presented with thyroid dyshormonogenesis and congenital hypothyroidism
The GTEx dataset in the HPA database (

Cntn6 KO led to impaired thyroid hormone biosynthesis and congenital hypothyroidism in mice. (
The Cntn6-KO mice exhibited a smaller body size and slower weight gain (Fig. 2B, C). At four months of age, compared with WT mice, Cntn6-KO mice had significantly lower serum triiodothyronine (T3) and thyroxine (T4) levels but higher serum TSH levels (Fig. 2D–F). In addition, the body weight and body length of Cntn6-KO mice were both significantly lower than those of WT littermates (Fig. 2G–J).
Their thyroid follicles formed normally, but the proportion of small follicles with the diameter <30 μm in Cntn6-KO mice was significantly higher than those in WT mice (Fig. 2K, L, O). Interestingly, the number of well-formed but function-impaired follicles that could not synthesize thyroid hormone was increased in Cntn6-KO mice (Fig. 2M, N). The above results suggested that Cntn6-KO mice had impaired thyroid hormone biosynthesis and exhibited a phenotype consistent with hypothyroidism.
Cntn6-KO downregulated the expression of crucial genes related to thyroid hormone biosynthesis
To further investigate the pathogenic mechanism through which Cntn6 deficiency leads to CH, we used quantitative PCR to assess the ontogeny of genes involved in thyroid function and development. Interestingly, there was no difference in the expression of genes related to thyroid development, while several crucial genes (Slc5a5, Tpo, and Duox2) involved in thyroid hormone biosynthesis were significantly suppressed in Cntn6-KO mice (Fig. 3A, B).
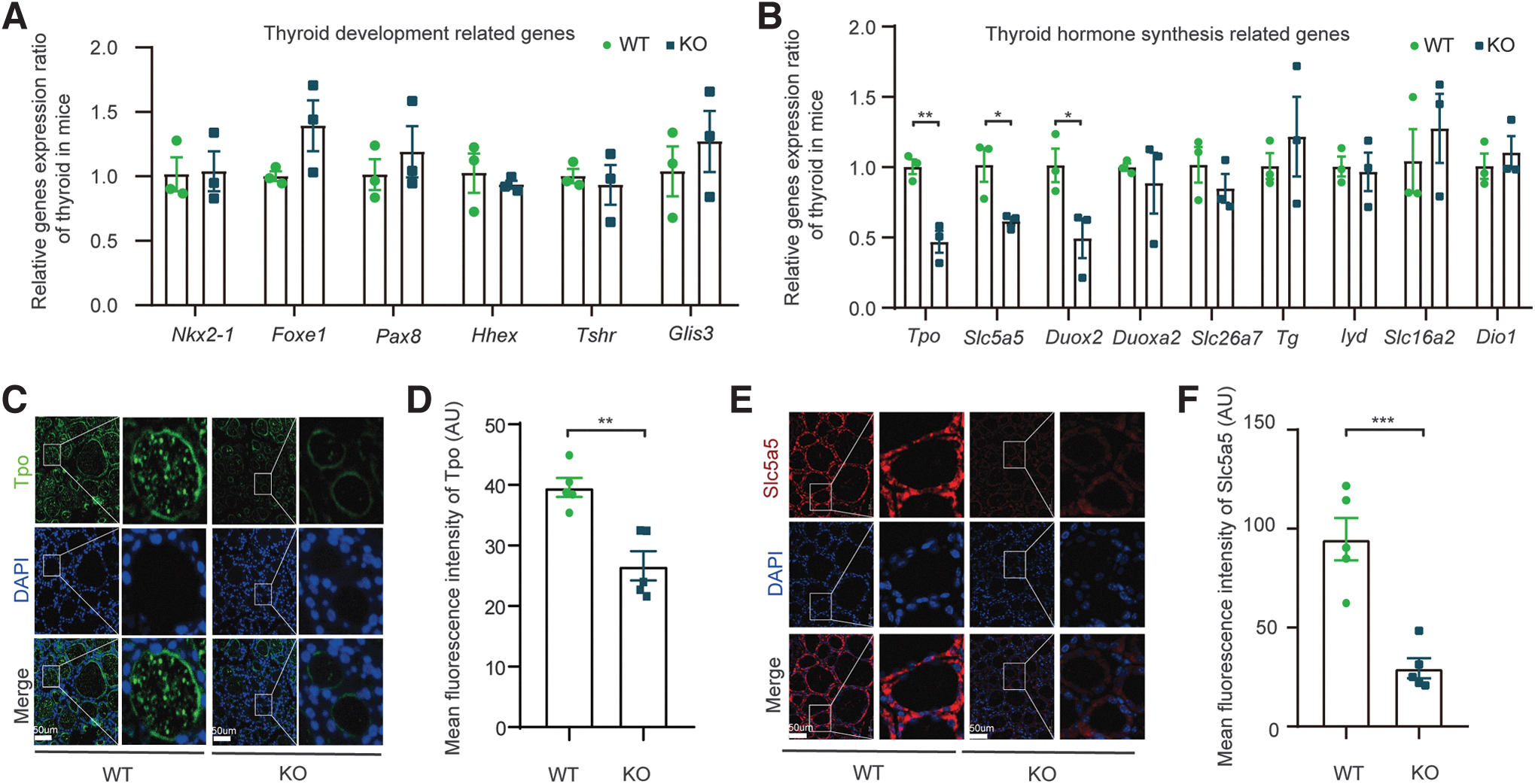
The expression of key genes involved in thyroid hormone biosynthesis was suppressed in Cntn6-KO mice. (
In thyroid tissue, SLC5A5 is responsible for the transport of iodide; and iodide uptake is typically the first step in thyroid hormone biosynthesis. 23 TPO oxidizes iodide ions to form iodine atoms for addition onto tyrosine residues on thyroglobulin, and catalyzes the coupling reaction of two iodized tyrosine residues, resulting in the formation of T3 or T4 in thyroglobulin. 24
In addition, the generation of H2O2 by DUOX2/DUOXA2 is essential for the production of thyroid hormone with TPO. 25 Subsequently, the significant downregulation of Tpo and Slc5a5 in the thyroid glands of Cntn6-KO mice was confirmed by immunofluorescence (Fig. 3C–F).
Pathogenic CNTN6 variants led to defective Notch signaling
We first assessed the binding affinity of WT and mutant CNTN6 with NOTCH1 through immunoprecipitation assays. The binding affinity between p.R217C and p.V879I variants with NOTCH1 significantly decreased, while the binding affinity between the p.T370I variant and NOTCH1 remained unchanged (Fig. 4A). Western blotting results demonstrated a decrease in the release of NICD from all three variants compared with the WT CNTN6 (Fig. 4B, C).
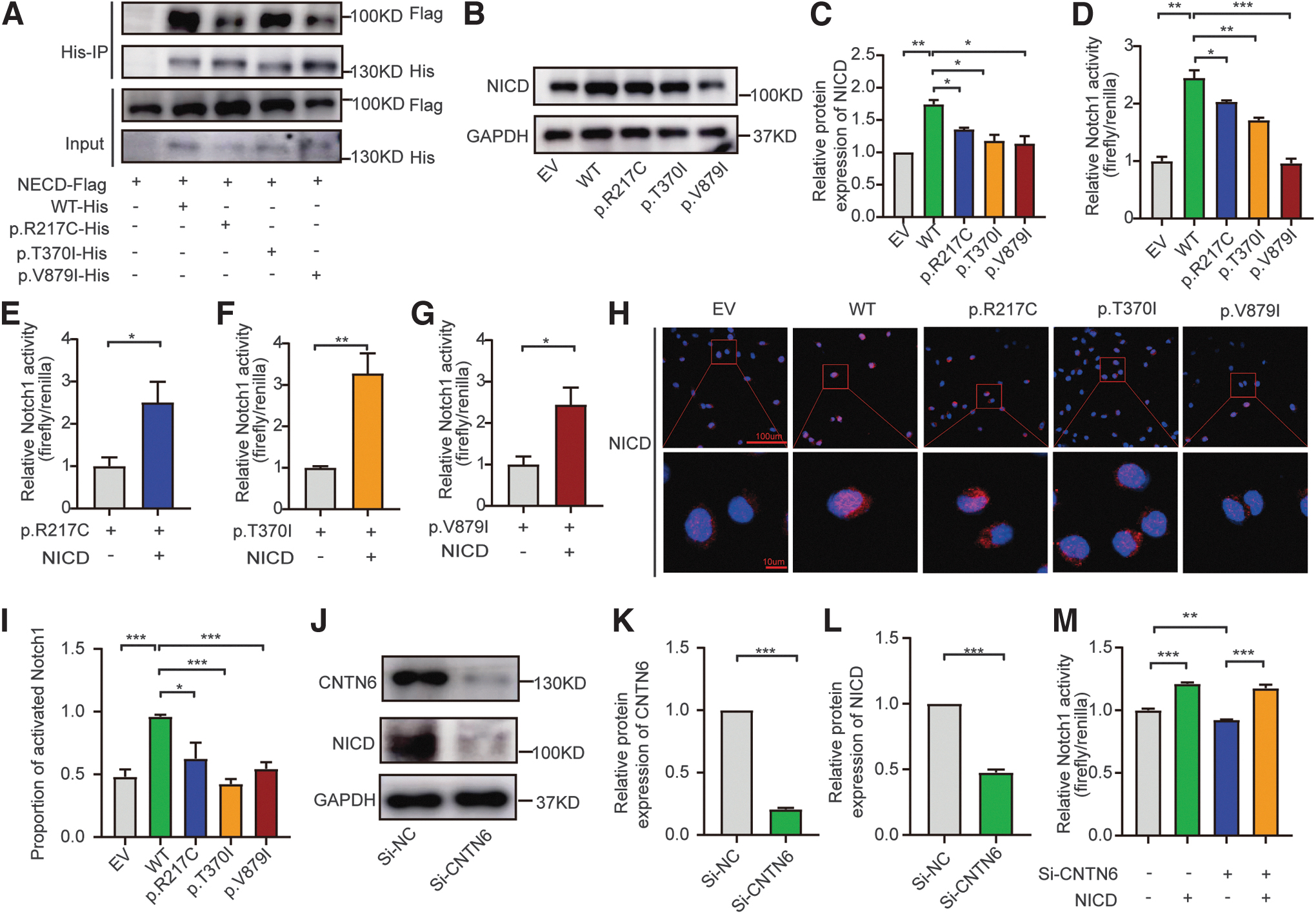
Pathogenic CNTN6 variants led to impaired Notch signaling. (
Next, we constructed a luciferase reporter plasmid with the NICD binding sites inserted upstream of the luciferase gene sequence to reflect NOTCH1 activity. 26 Similarly, all three variants inhibited the activity of luciferase, which could be rescued by co-transfecting with NICD (Fig. 4D–G). Further, all three variants resulted in a decreased nuclear translocation efficiency of NICD (Fig. 4H, I). Knockdown of CNTN6 by siRNA-4 also markedly reduced NICD expression levels and NOTCH1 activity in FTC-133 cells (Fig. 4J–M). The efficiency of the four siRNAs targeting CNTN6 is shown in Supplementary Figure S4. The sequence of siRNAs is listed in Supplementary Table S3.
CNTN6 variants suppressed the expression of genes associated with thyroid hormone biosynthesis through Notch signaling
Cntn6-KO in mice led to a downregulation in the expression of crucial genes (Slc5a5, Tpo, and Duox2) related to thyroid hormone biosynthesis, and CNTN6 variants resulted in impaired release of NICD and compromised Notch activity. Therefore, we speculated that CNTN6 variants influenced the expression of genes associated with thyroid hormone biosynthesis through Notch signaling.
We had identified NICD binding sites 27 in the SLC5A5, TPO, and DUOX2 promoter region and confirmed through CHIP-qPCR that NICD could bind to their promoter regions (Fig. 5A). All primers used for qPCR are shown in Supplementary Table S4. Consistent with our hypothesis, CNTN6 variants suppressed the mRNA expression of SLC5A5, TPO, and DUOX2 in HEK293T cells (Fig. 5B). Further, we confirmed that the protein expression levels of SLC5A5 and TPO were decreased in cells transfected with the CNTN6 mutant plasmids through Western blotting (Fig. 5C).
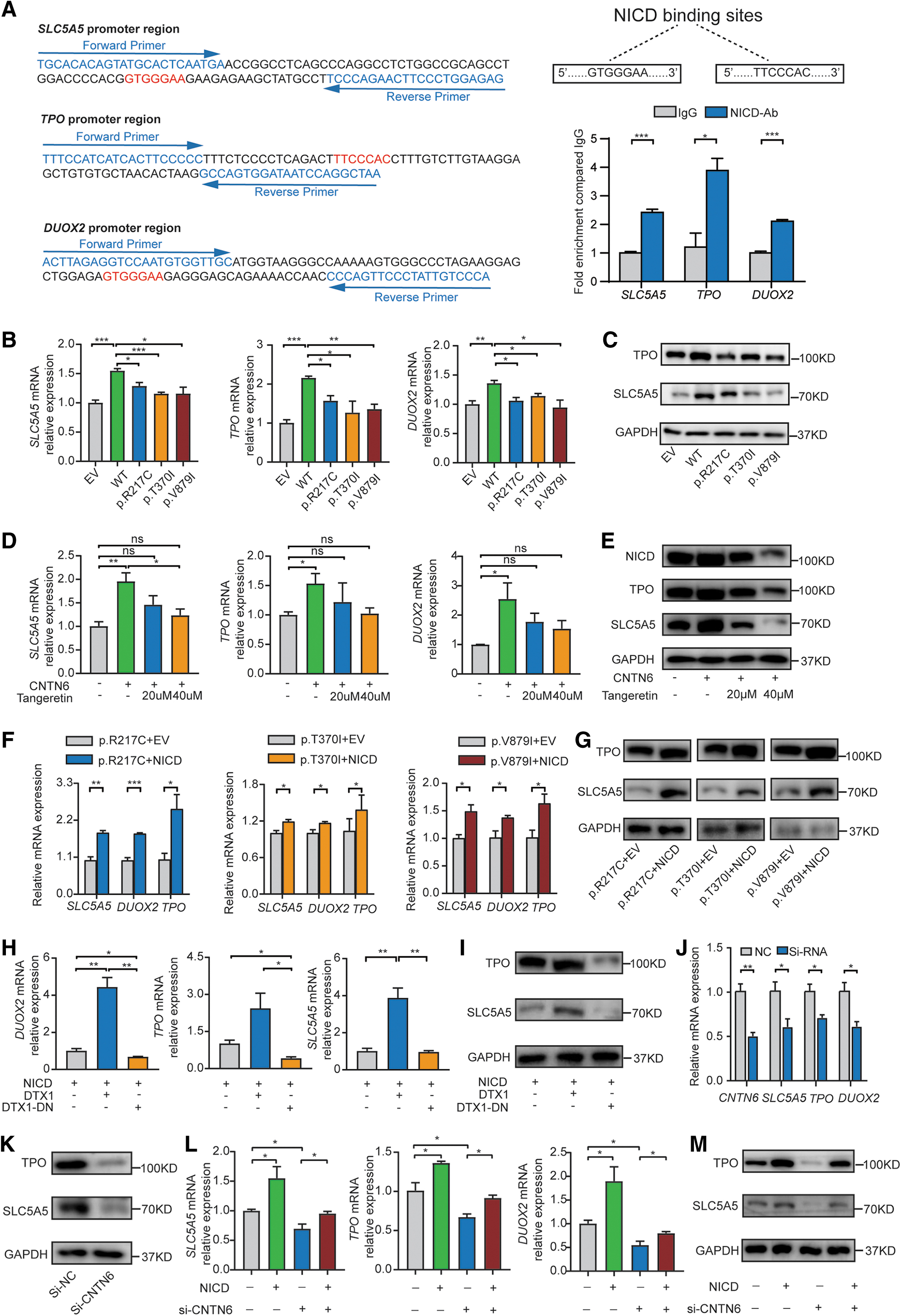
CNTN6 variants suppressed the expression of genes associated with thyroid hormone biosynthesis through Notch signaling. (
Unfortunately, we were unable to analyze the protein expression level of DUOX2 because there was no available DUOX2 antibody. Next, after treating the HEK293T cells with tangeretin, an inhibitor of NOTCH1, 28 the ability of CNTN6 to promote thyroid hormone biosynthesis was diminished (Fig. 5D, E). Notably, NICD over-expression rescued the downregulation of SLC5A5, TPO, and DUOX2 induced by the CNTN6 variants (Fig. 5F, G).
To further investigate whether DTX1 participates in CNTN6-induced thyroid hormone biosynthesis, HEK293T cells were transfected with pcDNA3.1-DTX1-Flag and pcDNA3.1-DTX1-dominant negative (DN)-Flag 29 plasmids. qPCR and Western blotting confirmed that DTX1 promoted the expression of SLC5A5, TPO, and DUOX2, whereas DTX1-DN inhibited their expression (Fig. 5H, I).
After CNTN6 was knocked down by siRNA-4 in FTC-133 cells, a decrease in the expression of SLC5A5, TPO, and DUOX2 was observed, which was differently restored by co-transfection with NICD (Fig. 5J–M).
Discussion
The etiology of CH, which is a sporadic disease with a genetic background, remains largely unknown. Known pathogenic genes of CH only account for 50% of cases, which prompt us to consider novel gene variants as the underlying cause. We identified three CNTN6 variants in two families with CH using whole exome sequencing. Through in vitro and in vivo experiments, we demonstrated that CNTN6 is a novel causative gene for CH, mediating thyroid hormone biosynthesis through Notch signaling.
CH can be divided into thyroid dysgenesis and thyroid dyshormonogenesis. Thyroid dysgenesis accounts for 85% of CH cases in the Caucasian population, which may manifest as athyreosis, ectopic gland, hypoplasia, or hemiagenesis. 30,31 However, thyroid dyshormonogenesis accounts for a greater proportion of CH cases in China. In this study, we recruited 599 patients with CH, 83% of whom presented with thyroid dyshormonogenesis accompanied by GIS, and 51.4% in whom the causative gene was not yet defined, which was consistent with the previous characterizations of the Chinese CH population. 7 –10
Among those patients with unknown pathogenic genes, three missense variants in CNTN6 were identified from two patients in two pedigrees. The transmission patterns of CNTN6 in two patients were consistent with autosomal recessive inheritance, which was the same pattern of inheritance as most of the thyroid dyshormonogenesis associated with genetic variants leading to CH, such as DUOX2, TPO, and SLC5A5. 9,23,32
Notably, the HPA database showed that CNTN6 exhibited high expression and tissue specificity in the thyroid and brain. However, studies on CNTN6 have been largely confined to the neurodevelopmental field. We demonstrated, for the first time, that Cntn6 was highly expressed in mouse adult thyroid tissue. Further, CH and thyroid dyshormonogenesis were found in humans with CNTN6 variants and in Cntn6-KO mice. In addition, Cntn6-KO in mice led to the downregulation of crucial genes (Slc5a5, Tpo, and Duox2) associated with thyroid hormone biosynthesis. These data indicated that normal thyroid hormone biosynthesis requires the functional CNTN6 protein.
The evolutionarily conserved Notch signaling pathways are further classified into canonical Notch signaling and non-canonical Notch signaling based on their dependence on different components (e.g., ligand and CSL). 33 Previous studies have shown that the canonical Notch signaling activated by the Jagged-1 ligand participates in the early phases of thyroid development, including primordium induction and budding, and the differentiation and proliferation of thyroid cells. 13,34,35
CNTN6 has been identified as a non-canonical ligand of NOTCH1, promoting oligodendrogliogenesis from progenitor cells and differentiation of oligodendrocyte precursor cells by activating the non-canonical Notch signaling. 15 However, the importance of this pathway in maintaining the normal function of the mature thyroid is currently unknown.
Our results indicated that all CNTN6 variants resulted in blocked NICD release and translocation into the nucleus, impaired NOTCH1 transcriptional activity, and decreased the expression of genes (SLC5A5, TPO, and DUOX2) related to thyroid hormone biosynthesis. The binding capacity of p.R217C and p.V879I variants to NOTCH1 was significantly reduced, and although the p.T370I variant was unchanged, its protein expression abundance was significantly reduced, both of which resulted in impaired NICD release and NOTCH1 activity (Fig. 6). These results suggested that all three variants of patient origin were loss-of-function variants.
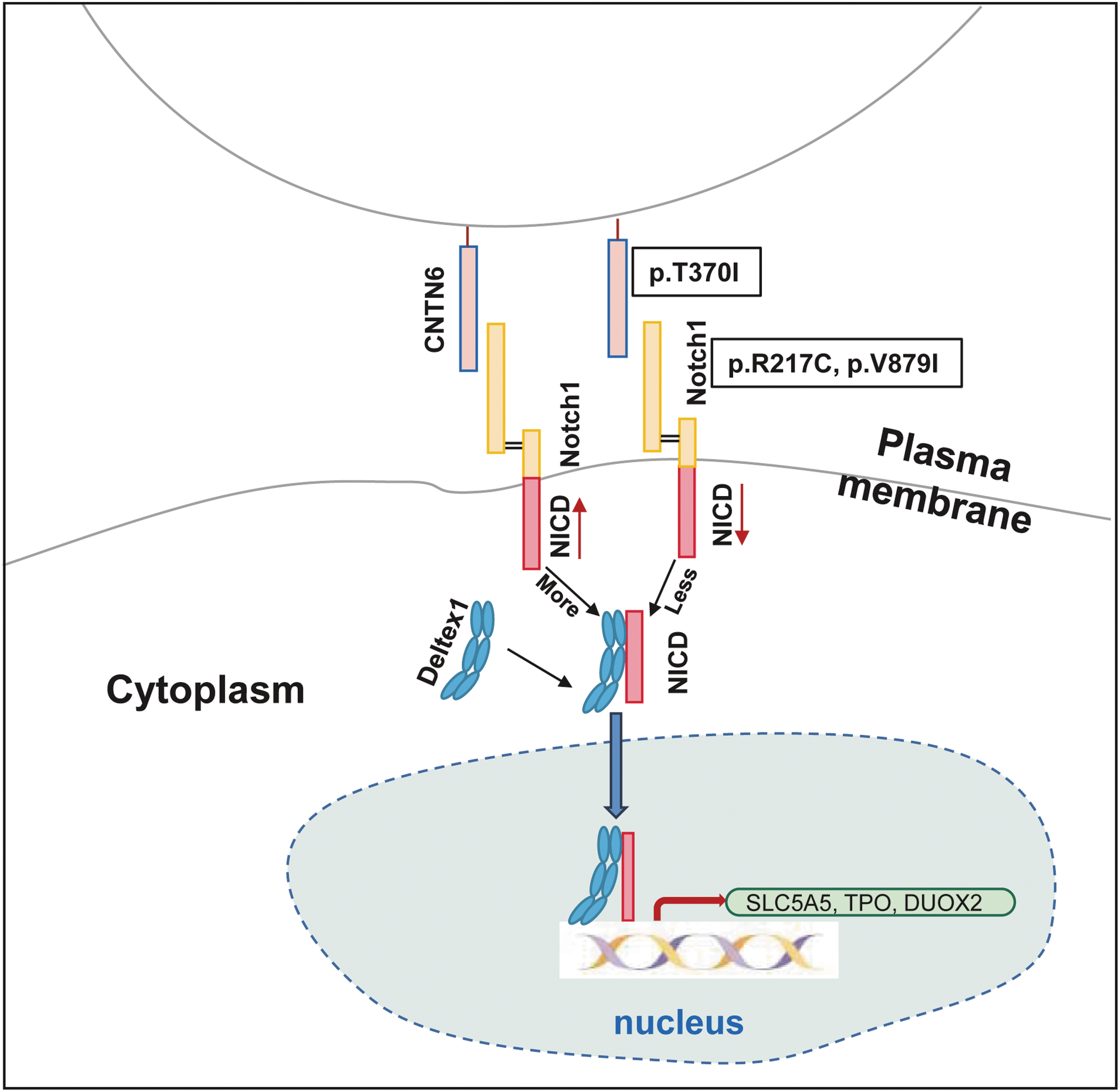
Mechanistic diagram of CNTN6 variants leading to thyroid dyshormonogenesis. The binding of WT CNTN6 to NOTCH1 leads to NOTCH1 activation and release of NICD, as well as recruitment of DTX1 into the nucleus. In the nucleus, NICD binds directly to the promoter regions of the SLC5A5, TPO, and DUOX2 genes, thereby promoting their expression. However, the binding capacity of CNTN6 p.R217C and p.V879I variants to NOTCH1 is significantly reduced, and although the CNTN6 p.T370I variant is unchanged, its protein expression abundance is significantly reduced, both of which result in impaired NICD release and entry to the nucleus. These further lead to a downregulation in thyroid hormone biosynthesis-related gene (SLC5A5, TPO, and DUOX2) expression, resulting in CH. NICD, NOTCH1 intracellular domain.
In mammals, after canonical ligand (Jagged or Delta-like) binding, the Notch receptor releases NICD, which is subsequently translocated into the nucleus and induces the transcription of HES1 through binding with DNA-binding protein CSL. 14 One study suggested that NOTCH1 in thyroid cancer cells significantly upregulated SLC5A5 expression by promoting the direct action of HES1 on the promoter region of SLC5A5. 11
In our study, we found that NICD activated and released by CNTN6 could bind directly to the promoter region of SLC5A5, TPO, and DUOX2 to facilitate their expression without the need for CSL and HES1 (Fig. 6), suggesting that CSL was not necessary in the non-canonical Notch pathway, consistent with a previous study. 36 Studies in Drosophila have identified DTX1 as encoding a positive regulator of the Notch signaling pathway. 37 Our study confirmed that positive regulation of DTX1 was required for CNTN6 to promote thyroid hormone biosynthesis through Notch signaling.
Several limitations should be considered when reviewing our study findings. First, although there was no difference in the expression of thyroid development-related genes in Cntn6-deficient mice, their thyroid follicle diameters were significantly decreased, and the effect of Cntn6 on thyroid development needs to be further investigated.
Second, CH can lead to intellectual disability if not treated in time. Clinical array comparative genomic hybridization testing and chromosome microarray platform analysis showed that CNTN6 copy number variants were associated with a wide spectrum of neurodevelopmental behavioral disorders, including intellectual disability. 21,22 As for the relationship between CNTN6 and CH and intellectual disability, this is a subject worthy of in-depth study in the future.
Taken together, our data indicated heretofore unsuspected roles for a member of the contactin family, CNTN6, in thyroid hormone biosynthesis. Three CNTN6 variants identified from two CH patients led to impaired thyroid hormone biosynthesis through the Notch signaling pathway under the regulation of DTX1. Thus, our work opens a whole new avenue for the future of the search for pathogenetic genes in CH patients and the development of personalized treatments for CH.
Footnotes
Acknowledgments
The authors would like to thank the patient participants of this study without whom this study would not be possible.
Authors' Contributions
All authors read and approved the final version of the manuscript. H.-Y.Z.: Conceptualization; Formal analysis; Writing—original draft; Visualization; and Validation. F.-Y.W.: Conceptualization; Formal analysis; and Visualization. C.-X.Z.: Validation; Visualization; and Data curation. C.-Y.W.: Formal analysis; Methodology; and Software. R.-J.C.: Data curation; Investigation. X.-Y.L.: Data curation. L.Y.: Resources. Y.Z.: Data curation. F.S.: Funding acquisition. F.C.: Investigation. R.-M.Y.: Data curation; Investigation, and Funding acquisition. H.-D.S.: Funding acquisition; Writing—review and editing. S.-X.Z.: Conceptualization; Funding acquisition; and Writing—review and editing.
Data Availability
All data and materials used in this study are available upon request.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the National Natural Science Foundation of China (Nos. 82301943, 82270826, 82200874, 82200873, 82170802, 82070816, and 81870537), Shanghai Municipal Education Commission Two-hundred Talent Program (No. 20161318).
Supplementary Material
Supplementary Materials
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4