
Abstract
Background:
Thyroid hormones regulate cardiac functions mainly through direct actions in the heart and by binding to the thyroid hormone receptor (TR) isoforms α1 and β. While the role of the most abundantly expressed isoform, TRα1, is widely studied and well characterized, the role of TRβ in regulating heart functions is still poorly understood, primarily due to the accompanying elevation of circulating thyroid hormone in TRβ knockout mice (TRβ-KO). However, their hyperthyroidism is ameliorated at thermoneutrality, which allows studying the role of TRβ without this confounding factor.
Methods:
Here, we noninvasively monitored heart rate in TRβ-KO mice over several days using radiotelemetry at different housing temperatures (22°C and 30°C) and upon 3,3′,5-triiodothyronine (T3) administration in comparison to wild-type animals.
Results:
TRβ-KO mice displayed normal average heart rate at both 22°C and 30°C with only minor changes in heart rate frequency distribution, which was confirmed by independent electrocardiogram recordings in freely-moving conscious mice. Parasympathetic nerve activity was, however, impaired in TRβ-KO mice at 22°C, and only partly rescued at 30°C. As expected, oral treatment with pharmacological doses of T3 at 30°C led to tachycardia in wild-types, accompanied by broader heart rate frequency distribution and increased heart weight. The TRβ-KO mice, in contrast, showed blunted tachycardia, as well as resistance to changes in heart rate frequency distribution and heart weight. At the molecular level, these observations were paralleled by a blunted cardiac mRNA induction of several important genes, including the pacemaker channels Hcn2 and Hcn4, as well as Kcna7.
Conclusions:
The phenotyping of TRβ-KO mice conducted at thermoneutrality allows novel insights on the role of TRβ in cardiac functions in the absence of the usual confounding hyperthyroidism. Even though TRβ is expressed at lower levels than TRα1 in the heart, our findings demonstrate an important role for this isoform in the cardiac response to thyroid hormones.
Introduction
It has been long recognized that thyroid hormones (THs) tightly control cardiac activity, with tachycardia as typical hallmarks of hyperthyroidism, and bradycardia observed in hypothyroidism. 1,2 THs, and specifically the active hormone 3,3′,5-triiodothyronine (T3), primarily act through regulating gene expression by two nuclear receptors, namely, TRα1 and TRβ, which show a different pattern of expression throughout the body. For the heart, studies suggest that TRα1 is the major isoform; however, TRβ also accounts for ∼30% of the total ligand-binding isoforms. 3 –5 Consequently, a number of studies demonstrated that mice carrying a mutation or deletion of TRα1 are bradycardic despite normal levels of THs. 6 –13 The role of TRβ in heart functions is more controversial, as a TRβ-KO leads to an accompanying hyperthyroid phenotype due to the disrupted negative feedback loop of the hypothalamus–pituitary–thyroid (HPT) axis, which complicates the interpretation of the phenotype. Nevertheless, some studies observed a modest increase by 6–11% in heart rate in TRβ-KO mice, which was attributed to the elevated TH levels and their action on cardiac TRα1. 7,9,14 Interestingly, however, previous research reported increased heart rate in TRα1-KO mice upon T3, indicating that TRβ might also play an important role in the regulation of cardiac functions. 12,13
Heart rate is also regulated by the autonomic nervous system (ANS), with the sympathetic branch increasing heart rate and the parasympathetic branch decreasing it. 15 One major factor that greatly affects autonomic activity in humans and rodents is ambient temperature. In mice, the usual housing temperature of ∼22°C leads to relatively high sympathetic activation, and even small deviations can significantly affect heart rate. 16 –19 Therefore, previous studies may have overestimated any cardiac phenotype due to the high sympathetic tone in mice housed at room temperature. To circumvent this issue, mice can be housed at thermoneutrality (30°C), a condition considered translationally more relevant. 20 –22 Even more importantly, housing at thermoneutrality ameliorates the hyperthyroidism of TRβ-KO mice, 23 thus advancing the field beyond the existing data, as it allows, for the first time, studying the role of TRβ in cardiac regulation without this confounding factor, and with an autonomic innervation that is more similar to humans and does not overestimate contributions of the sympathetic branch.
Here we used radiotelemetry in freely moving TRβ-KO mice to monitor heart rate at room temperature (22°C) and thermoneutrality (30°C). To dissect the state of the ANS, TRβ-KO mice were subjected to pharmacological denervation. Furthermore, to clarify the role of TRβ in T3-induced tachycardia and cardiac hypertrophy, heart rate and weight were measured upon oral T3 treatment. Finally, the expression of several cardiac genes involved in pacemaking, repolarization, and calcium handling was quantified.
Materials and Methods
Animal husbandry
Male TRβ-KO and control mice
24,25
were bred on the C57Bl/6NCrl background in the Gemeinsame Tierhaltung (GTH) of the University of Lübeck. Animals at ∼5 months were single housed in a 12-h light/12-h dark cycle (lights on at 6:00 am), temperature-controlled (22 and 30 ± 1°C) cabinet with ad libitum water and food (#1314, Altromin, Germany). Hyperthyroidism was induced at 30°C with 0.5 mg/L 3,3′,5-Triiodo-
In vivo phenotyping
Implantable radio transmitters (Mini-Mitter Respironics, Bend, OR, USA) were used to determine heart rate and locomotor activity. Radio transmitters were implanted as described previously 11,26,28,29 with recordings every 30 seconds. To study heart rate frequency distribution, data were split into 20-bpm (beats per minute) frequency bins. To study ANS contribution, mice were intraperitoneally injected first with scopolamine methyl bromide (0.1 mg/kg body weight; #S8502, Sigma-Aldrich, Germany) and 45 minutes later with timolol maleate (1 mg/kg body weight; #T6394, Sigma-Aldrich, Germany) in the first half of the inactive light phase. The differences between the median of each 45-min interval were calculated, and the intrinsic heart rate was recorded upon full receptor block 45 minutes after timolol administration. Electrocardiograms (ECGs) were recorded using ECGenie system (Mouse Specifics, Inc., MA, USA). Mice were acclimated to the electrode-fitted platform for at least 10 minutes before data collection.
T3, T4, and TSH determination
Total T3 (#DNOV053; NovaTec Immundiagnostica GmbH, Germany), T4 (#EIA-1781; DRG Instruments GmbH, Germany), and TSH (#MPTMAG-49K; Merck Millipore, Germany) levels were determined from serum by following manufacturer’s instructions.
Western blot analysis
Western blot was performed as described previously. 30,31 Hearts were homogenized in RIPA buffer with protease and phosphatase inhibitors. Proteins (25 µg) were separated on a 4–15% sodium dodecyl-sulfate polyacrylamide gel (Bio-Rad Laboratories, Germany), transferred onto a membrane (Merck Millipore, Germany), probed with a mouse anti-OxPhos antibody cocktail (1:2000 dilution; #45–8099, Thermo Fisher) or with a rabbit anti-SERCA2 antibody (1:1000 dilution; #4388, Cell Signaling), followed by a horseradish peroxidase-conjugated secondary immunglobuline G (IgG) antibody (1:5000 dilution; #P0447, Dako) or anti-rabbit-IgG antibody (1:5000 dilution; #P0448, Dako). The antigens were visualized using Clarity Max™ Western Substrate and ChemiDoc™ Touch Imaging System (Bio-Rad Laboratories, Germany) and normalized to total protein load.
In situ hybridization
The in situ hybridization for Trh mRNA was performed on PVN-containing brain slices (40 µm) using RNAscope® Multiplex Fluorescent Reagent Kit v2 (#436811-C2; ACD, USA) as described previously. 32 Brain slices were imaged using fluorescence microscope Leica DMI 6000B.
Reanalysis of Tabula Muris Senis single-cell data
Fully processed and annotated single-cell data for mouse heart were downloaded from Figshare (https://Figs.hare.com/ndownloader/files/23872838), and a Dot plot was generated for selected genes using Scanpy (v1.9.3). 33
Statistical analyses
Statistical analyses were performed using Excel (2016/2010/365, Version 2303) and GraphPad Prism 9.0 (GraphPad Software, US). All data are expressed as mean ± standard error of the mean (SEM), and differences were considered statistically different at p < 0.05. For comparison of two independent groups, Student’s t-test was used, while for comparisons with two variables such as temperature, genotype, and treatment, a two-way ANOVA was used. Further details on the specific tests can be found in Supplementary Table S2.
Results
To define the role of TRβ for heart rate, TRβ-KO and wild-type mice were noninvasively monitored using radiotelemetry at room temperature (22°C) and at thermoneutrality (30°C), revealing no difference between the genotypes at either condition (p = 0.83 for genotype at 22°C and p = 0.15 for 30°C, Fig. 1A and B). As expected, heart rate was reduced by thermoneutrality in both groups, indicating the expected shift from predominantly sympathetic nervous system (SNS) to more parasympathetic nervous system (PSNS) control. At 22°C, locomotor activity was reduced in TRβ-KO compared with wild-types, but solely due to a 23% reduced activity during the dark active phase (Fig. 1C, Supplementary Table S2), comparable to a previous study in a hypothalamic TRβ knockdown mouse. 34 This difference was no longer significant at 30°C (p = 0.11, Fig. 1D). Total T3 and T4 serum levels of TRβ-KO mice were elevated by 75% compared with controls at 22°C, but not at 30°C (Supplementary Fig. S1A, Supplementary Table S2), as expected from previous studies. 23,35 This was accompanied by high serum TSH at either temperature in the TRβ-KO mice (Supplementary Fig. S1A, Supplementary Table S2); however, Trh mRNA in the PVN, as well as pituitary Tshb and Dio2 mRNA expression, was also normalized at 30°C (Supplementary Fig. S1B and S1C, Supplementary Table S2). Together with reduced Trhr1 expression in the pituitary of TRβ-KO mice at 30°C (Supplementary Fig. S1B and S1C, Supplementary Table S2), this suggests that the hyperactivity of the HPT axis in TRβ-KO mice is dampened at all three levels.
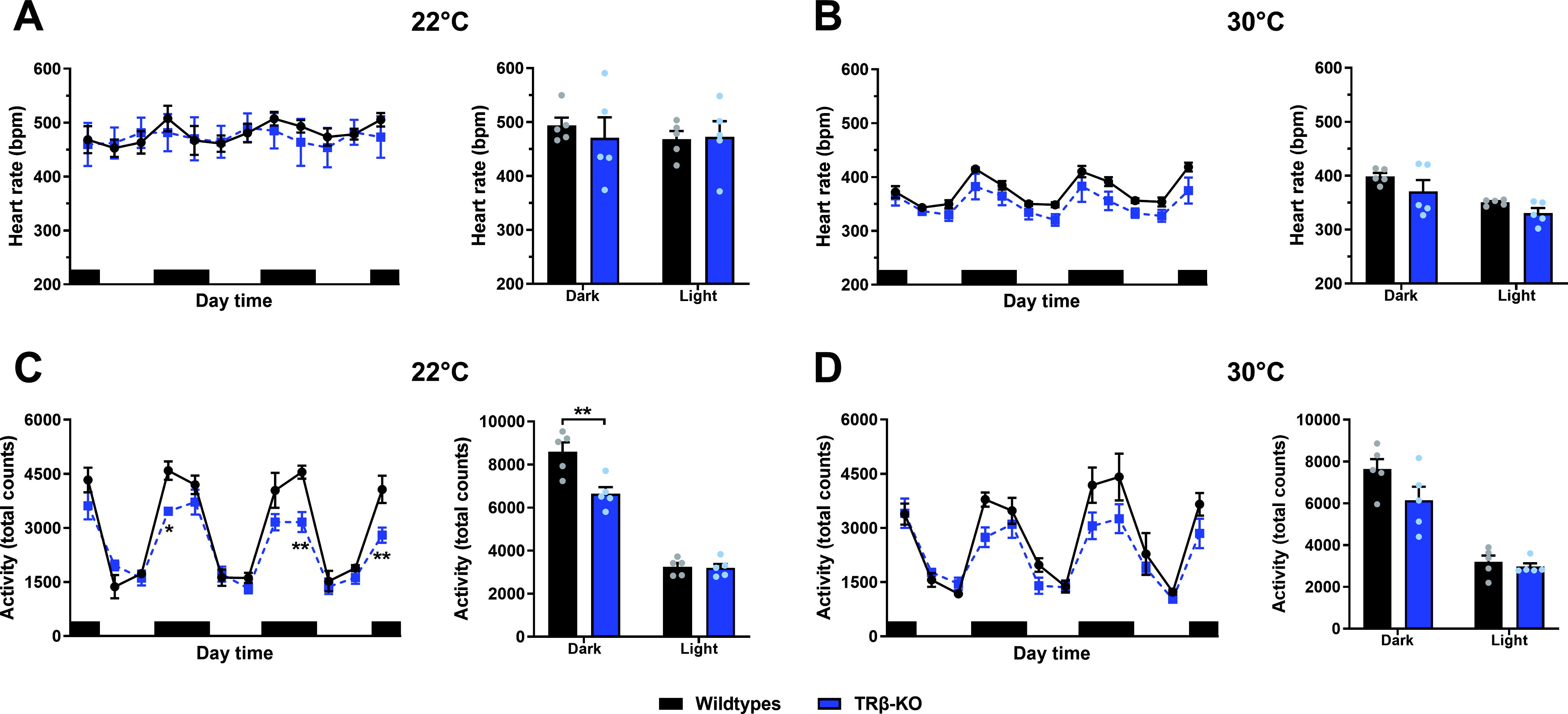
The effect of housing temperature on heart rate and locomotor activity in TRβ-KO mice.
To confirm the absence of tachycardia, we performed additional ECG recordings in another set of nonimplanted mice, which, despite significant shortening of QRS complex duration in TRβ-KO mice (p = 0.03, Supplementary Fig. S1D) confirmed similar heart rates in both genotypes (p = 0.48, Supplementary Fig. S1D and S1E). To quantify heart rate frequency distribution, a 3-day-long heart rate monitoring was split into 20-bpm bins, revealing that TRβ-KO mice were similar to wild-types at 22°C (p = 0.99, Fig. 2A). At 30°C, the predominant PSNS control was evidenced by a narrower distribution in both genotypes compared with 22°C (Fig. 2A, Supplementary Fig. S2A); however, heart rate frequency distribution was significantly narrower in TRβ-KO mice than in wild-types (Fig. 2A).
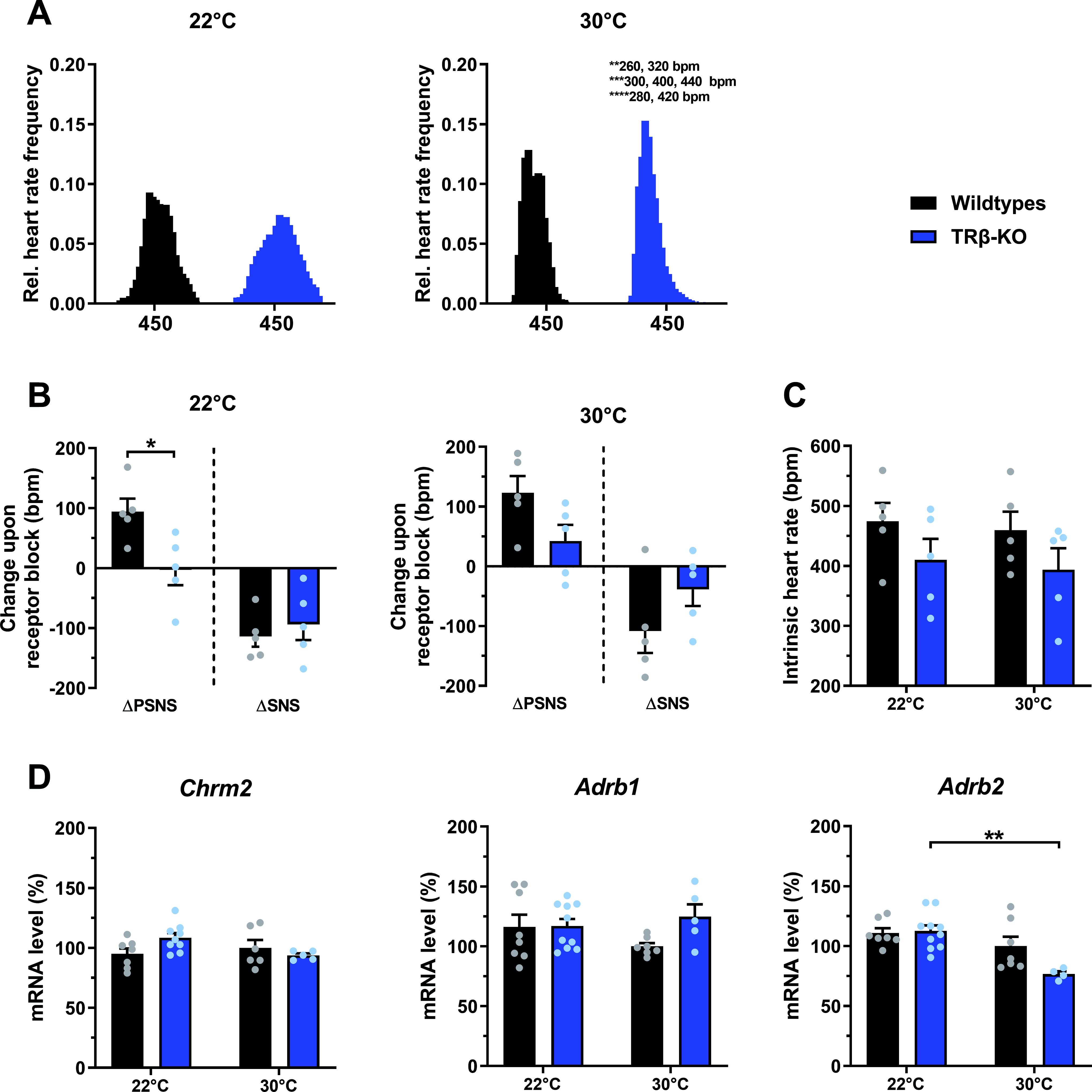
The effect of housing temperature on heart rate frequency distribution, pharmacological denervation, and cardiac mRNA receptor levels in TRβ-KO mice.
When we tested autonomic activity by pharmacological blockade of PSNS and SNS in vivo, we observed a PSNS impairment in TRβ-KO mice at 22°C (p = 0.02, Fig. 2B). This was partially rescued at 30°C, as the residual 66% reduction was no longer significant, suggesting a beneficial effect of thermoneutrality on PSNS activity in TRβ-KO (p = 0.07, Fig. 2B). SNS activity did not differ between groups at any housing temperature (Fig. 2B, Supplementary Table S2). Upon complete pharmacological autonomic receptor blockade, intrinsic heart rate was similar in TRβ-KO mice and controls at both temperatures (Fig. 2C, Supplementary Table S2). Likewise, cardiac mRNA expression of muscarinic receptor type 2 (Chrm2) and adrenoceptor type β1 (Adrb1), as a possible molecular mechanism underlying this pharmacological denervation response, was similar between groups, while Adrb2 was moderately reduced in 30°C TRβ-KO mice (Fig. 2D, Supplementary Table S2).
To induce tachycardia, we treated TRβ-KO and wild-type mice with T3 in the drinking water for 12 days at 30°C. As previously shown, 6,36 this pharmacological treatment led to a robust elevation in serum T3 and a parallel suppression of T4, with somewhat higher T3 levels in TRβ-KO mice (p = 0.03, Supplementary Fig. S2B). While T3 treatment prominently increased heart rate in wild-types already after 2–3 days as expected, this effect was blunted in TRβ-KO mice resulting in a significantly lower heart rate compared to wild-types (p < 0.0001 for interaction, Fig. 3A). In addition, while the minimum heart rate during the inactive light phase was unaltered by T3 treatment, the maximum heart rate in the active dark phase was reduced in TRβ-KO compared with controls (Supplementary Fig. S2C, Supplementary Table S2), leading to a reduced delta heart rate in TRβ-KO mice (p = 0.01, Fig. 3B). As reported previously, 6 the heart rate frequency distribution of wild-types broadened upon T3, whereas that of TRβ-KO mice remained narrower (Fig. 3C and Supplementary Fig. S2D and E, Supplementary Table S2). While heart weight was not different between untreated groups at 30°C, T3 treatment resulted in a significant increase in wild-type but not TRβ-KO heart weight, resulting in a significant 14% difference compared with controls (Fig. 3D, Supplementary Table S2). On the molecular level, this was supported by impaired T3 induction of Nppb and Col1a1 (Fig. 3E, Supplementary Table S2).

The effect of T3 treatment on heart rate, heart rate frequency distribution, heart weight, and gene expression in TRβ-KO mice at 30°C.
In line with the radiotelemetry data, mRNA levels of the two pacemaker genes Hcn2 and Hcn4 were comparable in untreated TRβ-KO and controls. While the expression of Hcn2 was strongly induced by T3 in both genotypes, this occurred to a much lesser extent in TRβ-KO mice (+408% vs. 132%), resulting in significantly lower mRNA levels compared with T3-treated wild-types. Likewise, the expression of Hcn4 was significantly induced only in wild-type animals (Fig. 4A, Supplementary Table S2). The qPCR analysis of potassium channels implicated in cardiac repolarization revealed that Kcna7 was significantly induced by T3 treatment in wild-type but not in TRβ-KO mice. While Kcnj3 and Kcnq1 were significantly reduced by T3 treatment in both genotypes, Kcnh2 was unresponsive to T3 and significantly lower in TRβ-KO mice compared with wild-types, suggesting that regulation of this channel requires intact TRβ (Fig. 4A, Supplementary Table S2). The ratio between Myh6 and Myh7 as a measure of cardiac hypertrophy showed the expected significant elevation in both groups (Fig. 4A, Supplementary Table S2). Regarding genes involved in calcium handling and cardiac contraction, T3 slightly but significantly lowered Atp2a2 (Serca2) mRNA levels only in wild-type mice, while Pln, an endogenous inhibitor of SERCA2 activity, was significantly decreased in both genotypes (Fig. 4A and Supplementary Fig. S3A, Supplementary Table S2). On the protein level, however, SERCA2 was not altered (Supplementary Fig. S3B, Supplementary Table S2). The basal mRNA levels of Ryr2 were significantly increased by 30% compared with controls, and, interestingly, while T3 treatment enhanced Ryr2 in wild-types, it was decreased in T3 treated TRβ-KO mice. However, there were no significant differences between wild-types and TRβ-KO upon T3 in any of these calcium handling–related genes (Fig. 4A, Supplementary Table S2), suggesting that they are not involved in the observed partial resistance. We observed minor reductions in Chrm2 and Adrb2 in TRβ-KO mice at 30°C, while Adrb1 was not affected (Supplementary Fig. S3A, Supplementary Table S2). Interestingly, despite its low expression, we observed a clear TRβ-dependent acute regulation of Dio2 (Supplementary Fig. S3A, Supplementary Table S2). To test for major metabolic alterations in cardiac respiration, we analyzed the five complexes of the oxidative phosphorylation chain on protein level, which were, however, not affected by genotype or treatment (Supplementary Fig. S3C, Supplementary Table S2).

Cardiac gene expression with and without T3 treatment in TRβ-KO mice at 30°C.
Finally, to better understand whether TRβ could be directly involved in the regulation of the tested genes, we reanalyzed published single-cell RNA sequencing data of adult mouse hearts to identify TRβ expressing cell types using the Tabula Muris Senis data. 37 These data showed expression of TRβ primarily in cardiomyocytes, as well as lower TRβ expression in endocardial cells, smooth muscle cells, and fibroblasts, which also expressed Dio2 (Fig. 4B), thus supporting the possibility of a TRβ dependent regulation.
Discussion
The main findings of the present study were as follows: TRβ-KO mice showed (1) normal heart rate both at room temperature and at thermoneutrality, (2) moderately reduced locomotion and parasympathetic activity at room temperature, which were partially rescued at thermoneutrality; (3) resistance to T3-induced tachycardia and cardiac hypertrophy, and (4) altered expression of several cardiac genes, including Hcn2, Kcna7, and Ryr2. While these findings suggest only a negligible role for TRβ under baseline conditions, the role of the receptor might be more relevant for developing the symptoms of systemic hyperthyroidism.
TRβ is required for T3 induced cardiac hypertrophy
The induction of cardiac hypertrophy is one of the most classic effects of THs, 38 which, however, seem to require the ANS. 39 In fact, THs have been reported to increase the expression/activity of β-adrenoceptors leading to a greater sensitivity of the heart to sympathetic stimulation and, eventually, to positive inotropic effects, 40 –42 as chronic administration of the β-adrenoceptor agonist isoproterenol also increases heart weight. 43,44 Importantly, the β-adrenoceptor blocker propranolol inhibits T3-induced cardiac hypertrophy and tachycardia. 45 Corroborating these findings, cardiac hypertrophy after pharmacological treatment with T3 was not observed in mice lacking all the β-adrenoceptors. 46 In addition, Ortiga-Carvalho et al. 47 observed that while whole-body TRβ mutant mice developed cardiac hypertrophy upon T3 treatment, this effect was not displayed by animals with cardiac-specific TRβ mutation, clearly demonstrating the necessity of extra-cardiac TH actions for the development of hypertrophy.
In the present study, we found that T3 treatment of control animals led to a 36% increase in heart weight, a clear indication of cardiac hypertrophy. This effect is not surprising considering that we observed a five- to sevenfold elevation in circulating T3 6,36 and that even lower doses of T3 can cause a significant increase in heart weight. 26 This observation was accompanied by induced Nppb and Col1a1 cardiac expression upon T3 treatment, indicative of increased cardiomyocyte size and collagen deposition. 48 In contrast, TRβ-KO mice show no increase in heart weight or in Nppb and Col1a1 levels upon T3, demonstrating that TRβ is required for T3-induced cardiac hypertrophy at 30°C, an effect previously observed at room temperature. 47,49,50 It remains at present unclear whether this is the result of altered acute TRβ action in other tissues such as the brain or whether permanent defects arising from the lack of TRβ during cardiac development could also be involved.
Lack of TRβ does not affect basal heart rate frequency at 22°C and 30°C
Previous studies have shown a modest increase in heart rate of TRβ-KO mice as a result of their higher TH levels; however, these experiments were all conducted at room temperature, thus permanently exposing the animals to minor cold stress. 7,9,14 Our in vivo radiotelemetry and ECG data now show that the lack of TRβ has no effect on heart rate at room temperature, indicating that the moderate ∼75% elevation in TH levels at 22°C might not be sufficient to induce tachycardia, corroborating previous results showing a normal heart rate frequency profile in wild-type mice with similarly elevated TH levels. 26
A particular advantage of our present study is the phenotyping at thermoneutrality, a condition that better resembles that of humans, as animals are not cold-stressed and heart rate is predominantly under parasympathetic control. 21,22 More importantly, housing at 30°C leads to a significant normalization of TH levels in TRβ-KO mice, allowing to assess the role of TRβ in the heart without the confounding hyperthyroidism. Our in vivo data at thermoneutrality now show normal heart rate in untreated TRβ-KO mice together with comparable cardiac expression of Hcn2 and Hcn4, supporting the notion that at baseline these two pacemaker genes are mainly regulated by TRα1, as observed previously. 4,51 Upon T3, our TRβ-KO mice developed tachycardia at 30°C, but not to the same extent as the control animals, suggesting that TRβ plays an important role in reaching maximum heart rate in a hyperthyroid state, potentially by providing a higher number of TRs per cell for full gene regulation. Our data are in agreement with a previous study showing failure of TRβ-KO mice to develop tachycardia upon T3 treatment; 7 however, although the T3 dose was comparable, the treatment was restricted to four days, which may be insufficient to generate tachycardia in the somewhat resistant TRβ-KO mice. Most importantly, a robust increase in heart rate was observed in TRα1-KO mice upon T3, demonstrating that TRβ can in fact contribute to T3-induced tachycardia under certain conditions. 12,13
On the molecular level, our data of blunted tachycardia in T3-treated TRβ-KO mice match a reduced induction by T3 of the pacemaker gene Hcn2, as well as the potassium channel Kcna7. It remains to be determined whether this constitutes a developmental defect in TRβ-KO mice similar to that observed in TRα1 mutant mice 6,27 or whether lack of TRβ impairs, for example, other tissues such as the central adrenergic system, which is crucial for pacemaker gene induction. 46
Altered PSNS activity and heart rate distribution in TRβ-KO mice
Since heart rate is also regulated through the ANS, we also studied SNS and PSNS. Interestingly, while SNS activity was normal at both temperatures, TRβ-KO mice displayed reduced PSNS activity at 22°C, which was partially rescued by thermoneutrality. This improvement in PSNS activity was not due to changes in muscarinic (Chrm2) and/or β-adrenergic (Adrb1) receptors. Importantly, our observation of normal intrinsic heart rate indicates no cardiac defects caused by the lack of TRβ, suggesting that developmental and/or functional defects may reside in other structures (e.g., the hypothalamus). At present, it is difficult to establish whether the decrease in PSNS activity is related to the T3 levels at 22°C and 30°C. While we observed previously that a sixfold increase in T3 levels leads to decreased PSNS activity in mice housed at thermoneutrality, 6 wild-type mice housed at room temperature with moderately increased T3 levels similar to those of the TRβ-KO mice had normal PSNS activity, 26 which suggests that T3 only affects the PSNS above a certain threshold. Given the normal SNS and PSNS activity in TRβ KO mice at room temperature, 7 it seems likely that the effects of TRβ on PSNS activity are negligible unless they become severely hyperthyroid.
Interestingly, heart rate frequency distribution was generally broader at 22°C and narrower at 30°C, indicative of a less stringent central control of heart rate at room temperature. In fact, while wild-type mice show broader heart rate frequency distribution during T3 treatment as expected, 6 TRβ-KO mice remained permanently narrow, suggesting that TRβ mediates heart rate stability in response to THs. This could be caused by developmental defects occurring in the central nervous system, as TRβ-KO mice show a ∼40% reduction in anterior hypothalamic parvalbumin neurons, known to orchestrate autonomic, cardiovascular, and stress response/functions. 11,52,53
Conclusions
Taken together, the present findings point toward a role of TRβ in regulating frequency distribution rather than average heart rate by a yet unknown mechanism possibly involving tissues other than the heart. More importantly, TRβ seems to be required for the full development of T3-induced tachycardia and hypertrophy; however, it remains unclear whether this is an acute effect or the consequence of the lack of TRβ actions during development. Likewise, it remains to be established whether the same effects can also be observed in female mice and whether nongenomic actions in particular in angiogenesis could contribute. 54 Here, a cardiomyocyte-specific mutation of TRβ will be required to dissect the individual cellular contributions to the observed phenotype without indirect effects through other tissues—ideally an inducible system to avoid confounding developmental effects as observed previously for TRα1. 6
That the hearts of TRβ-KO animals retain most of their sensitivity to TH is, however, clinically important in the light of recent findings of increased cardiovascular morbidity and mortality in RTHβ patients. 55 In this study, it may also be of relevance that the hyperthyroidism of TRβ-KO mice depends on environmental temperature, which could suggest that cardiovascular issues in resistance to TH (RTHβ) patients 55 may be more pronounced in the cold. Finally, any drug developed to target TRβ signaling in patients, such as resmetirom, 56 needs a high liver specificity to avoid unintentional activation of cardiac TRβ.
Footnotes
Acknowledgments
Authors’ Contributions
R.D., S.C.S., and J.M. conceptualized the study; R.D., S.C.S., K.J., N.L.-A., and A.C. performed the experiments; J.R. performed qPCR; R.D., B.O., R.O., and J.M. analyzed the data; R.D., H.M.-F., L.C.M., D.F., B.O., R.O., M.S., and J.M. interpreted the data; R.D. and J.M. drafted the article. All authors provided critical revision of the article and approved its final version for publication.
Author Disclosure Statement
The authors have nothing to disclose.
Funding Information
The authors are grateful for funding from the German Research Council DFG in the framework of CRC/TR 296 “LOCOTACT” (funding ID 424957847), GRK1957 “Adipocyte Brain Crosstalk,” and MI1242/3-2.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Table S1
Supplementary Table S2