
Abstract
Background:
Stimulation of ventricular hypertrophy and heart rate are two major cardiac effects of thyroid hormone (TH). The aim of this study was to determine in vivo which TH receptor (TR)—α or β—and which mode of TR action—canonical gene expression or DNA-binding independent noncanonical action—mediate these effects.
Methods:
We compared global TRα and TRβ knockout mice (TRαKO; TRβKO) with wild-type (WT) mice to determine the TR isoform responsible for T3 effects. The relevance of TR DNA binding was studied in mice with a mutation in the DNA-binding domain that selectively abrogates DNA binding and canonical TR action (TRαGS; TRβGS). Hearts were studied with echocardiography at baseline and after 7 weeks of T3 treatment. Gene expression was measured with real-time polymerase chain reaction. Heart rate was recorded with radiotelemetry transmitters for 7 weeks in untreated, hypothyroid, and T3-treated mice.
Results:
T3 induced ventricular hypertrophy in WT and TRβKO mice, but not in TRαKO mice. Hypertrophy was also induced in TRαGS mice. Thus, hypertrophy is mostly mediated by noncanonical TRα action. Similarly, repression of Mhy7 occurred in WT and TRαGS mice. Basal heart rate was largely dependent on canonical TRα action. But responsiveness to hypothyroidism and T3 treatment as well as expression of pacemaker gene Hcn2 were still preserved in TRαKO mice, demonstrating that TRβ could compensate for absence of TRα.
Conclusions:
T3-induced cardiac hypertrophy could be attributed to noncanonical TRα action, whereas heart rate regulation was mediated by canonical TRα action. TRβ could substitute for canonical but not noncanonical TRα action.
Introduction
The heart is a major target organ of thyroid hormones (TH) and especially heart rate and ventricular growth are stimulated by TH. TH effects are mediated by the TH receptors (TR) TRα1 and TRβ1 and 2. 1 –3 TRs act as ligand-dependent transcription factors regulating the expression of TH-target genes by binding to TH response elements (TRE) on the DNA (type 1 TR signaling, canonical). In addition, T3 (3,3′,5-triiodo-l-thyronine) and TRs mediate activation of signaling pathways, e.g., (phosphoinositide 3-kinase) PI3K/Akt and (mitogen-activated protein kinase) MAPK/ (extracellular signal-regulated kinase) ERK (type 3 TR signaling, noncanonical). 4 –11 This mode of action is independent from DNA binding of TRs. The precise mechanisms of TH action leading to different TH-mediated cardiac effects is still unclear and requires the determination of the responsible TR isoform, TRα or TRβ, and of the mode of TR action, canonical or noncanonical. TH excess, either hyperthyroidism in patients or TH treatment of mice, leads to cardiac hypertrophy through a combination of TH action in cardiomyocytes, including regulation of gene expression (e.g., Myh6 and 7, adrenergic receptors) and regulatory pathway activation (e.g., PI3K and MAPK signaling), and interaction with other signaling systems, especially the sympathetic nervous system. 12 Although TRα is the predominant TR isoform in the heart, T3-induced ventricular growth has not been unanimously attributed to a TR isoform. For instance, T3 induced cardiac hypertrophy only in WT but not in mice expressing a dominant-negative TRβΔ337T mutant receptor. 13 Of course, the dominant negative effect of TRβΔ337T could have masked TRα-mediated effect. Another study suggested a role of TRβ in cardiac hypertrophy. Thyroxine (T4) treatment of hypothyroid mice for 4 weeks appeared to lead to an increase in ventricular mass, calculated from echocardiography, in wild-type (WT) and TRα knockout (TRαKO) mice, whereas absence of TRβ in TRβ knockout (TRβKO) mice prevented the T4-induced increase in ventricular mass. 14 However, ventricular weights were not normalized to tibia length and the actual ventricular weights, measured with a scale after organ collection, seemed not to be different between the genotypes, precluding firm conclusions from this study regarding the role of TR isoforms in ventricular growth. In vitro studies attributed cardiomyocyte hypertrophy to TRα1 because total cardiomyocyte protein content in cardiomyocytes, a surrogate parameter for hypertrophy, correlated with TRα1 signaling and not with TRβ. 15,16 The role of TR isoforms in TH-induced cardiac hypertrophy in vivo remained controversial.
Interestingly, T4 treatment in rats increased heart weight and Akt phosphorylation, indicating activation of PI3K by TH. 17 In mice, T4 treatment induced cardiac hypertrophy, which was abrogated by cotreatment with rapamycin, an inhibitor of mammalian target of rapamycin (mTOR). 18 In vitro, T3 rapidly increased PI3K activity and subsequently Akt and mTOR phosphorylation in rat cardiomyocytes. 19 In addition, TRα and the regulatory PI3K subunit p85α formed complexes in cardiomyocytes. Together, these data suggested that ventricular growth in response to TH could at least in part be mediated by noncanonical TR signaling.
Here we studied genetic mouse models to determine which TR—TRα or TRβ—and which mechanism—canonical or noncanonical signaling—mediates TH-induced cardiac effects such as ventricular growth or heart rate regulation. We compared global TRα knockout mice and TRβ knockout mice with WT mice to determine the TR isoform responsible for the T3 effects on the cardiac hypertrophy and heart rate. To determine the relevance of TRs’ DNA binding and, therefore, the potential relevance of noncanonical TR signaling, we studied mice with mutations in the TR DNA-binding P-box, changing glutamic acid (E) and glycine (G) to glycine and serine (S) (EG to GS, codons 71/72 in Thra and 125/126 in Thrb, respectively). 7 These mutations abrogate the TRs’ DNA binding and canonical actions in TRαGS and TRβGS mice. A comparison of WT, TRαKO and TRβKO, and TRαGS or TRβGS mice allows to attribute TH effects to the relevant TR isoform and to the underlying TR signaling mechanism. The results demonstrated that T3-induced ventricular growth was mediated by noncanonical TRα action and heart rate was regulated by canonical TRα action with contribution of TRβ.
Materials and Methods
Animal studies
All animal experiments were approved by the Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen (LANUV-NRW, AZ: 84-02.04.2017.A157) and performed in accordance with the German regulations for Laboratory Animal Science and the European Health Law of the Federation of Laboratory Animal Science Associations. TRα knockout (TRα0/0) and TRβ knockout (TRβ−/−) mice, here referred to as TRαKO and TRβKO, respectively, were obtained from the European Mouse Mutant Archive (https://www.infrafrontier.eu). 2,3 Generation of TRαGS and TRβGS mutant mice was described previously. 7 Mice were housed in the central animal facility at the University Hospital Essen in individually ventilated cages at 21 ± 1°C in an alternating 12:12-hour light-dark cycle and fed standard chow (Sniff, Soest, Germany) and tap water provided ad libitum. For echocardiography studies, T3 was administered orally via drinking water containing 400 ng/mL T3 (Sigma-Aldrich, USA) for 7 weeks. Echocardiography measurements were performed with male mice aged approximately 8 weeks (echo 1) and 7 weeks later (echo 2) (Fig. 1A). Mice were anesthetized with pentobarbital (20 mg/g body weight) and examined with a Vevo2100 (VisualSonics) echocardiography device. Fractional shortening, end-diastolic interventricular septum thickness (IVS;d), left ventricular posterior wall thickness (LVPW;d), and left ventricular inner dimension (LVID;d) were recorded and calculated. 20,21
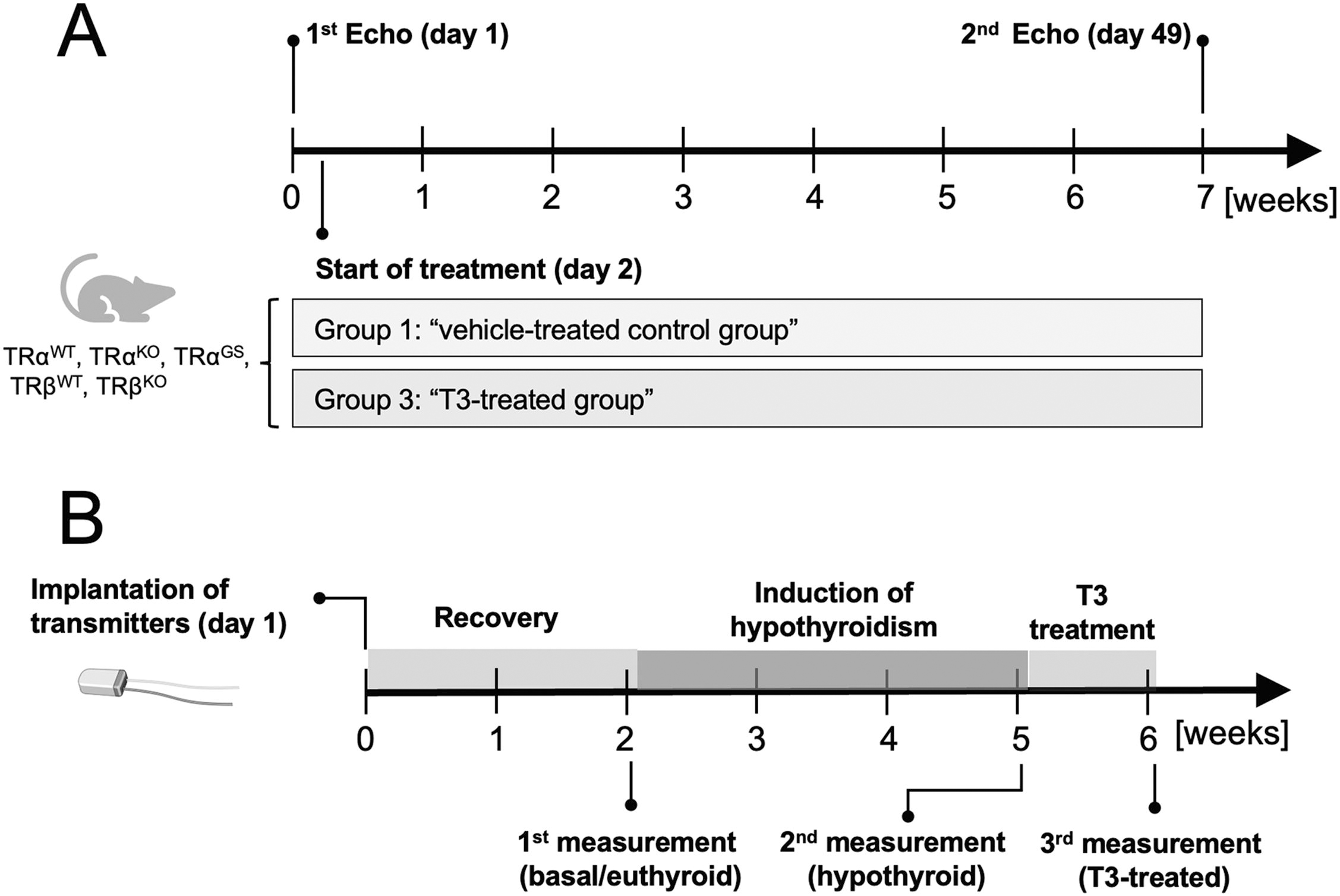
Echocardiography and radio telemetry study design.
As part of the systemic phenotyping, electrocardiography (ECG) was performed at the German Mouse Clinic (GMC) in Munich in awake male 15-week-old untreated WT, TRαKO, TRαGS, TRβKO, and TRβGS mice. 22,23 Mice were maintained in individually ventilated cages with water and standard mouse chow according to the directive 2010/63/EU, German laws and GMC housing conditions (www.mouseclinic.de). All tests were approved by the responsible authority of the district government of Upper Bavaria.
For radio telemetry experiments, chronic hypothyroidism was induced by feeding male mice low iodine food (LID; TD.95007, Harlan Laboratories) and supplementing the drinking water with 0.04% methimazole, 0.5% sodium perchlorate (ClO4 −), and 0.3% saccharine as sweetener for 3 weeks. High T3 serum concentrations were achieved by adding 400 ng/mL T3 to drinking water for 6 days.
Long-term in vivo heart rate was measured with a radio telemetry system (Data Science International). 24 Transmitters (DSI PhysioTel® Transmitter [ETA-F10]) were implanted in 4–5-month-old mice with a minimal body weight of 25 g. Mice were anesthetized with fully antagonizable anesthesia followed by a subcutaneous injection of carprofen (5 mg/kg BW). After placing the transmitter into the abdominal cavity, the anode was tunneled subcutaneously to the neck, where the loop was attached to a muscle with surgical thread. The cathode was attached to a muscle below the heart so that both electrodes formed a diagonal line over the heart. Following surgery, mice were woken up with the anesthesia antagonist and supplied with 15% glucose solution as drinking water and steeped food pellets. Mice were allowed to recover for 10–14 days and were treated with analgesics for 4 days. After the recovery period, heart rate was constantly recorded for 3 days each in untreated mice and after treating mice with hypothyroidism-inducing food and drinking water for 3 weeks (Fig. 1B). Next, T3 was added to the drinking water and heart rate was recorded for 6 days to investigate the transition from hypothyroidism to early (day 1–3) as well as late T3 treatment phase (day 4–6). For analysis of the recorded data, they were assigned to the natural active and inactive state of the animals. Mean values were saved every 30 seconds.
Organ collection
Mice were sacrificed by cervical dislocation and body weight was recorded. Blood was collected by heart puncture and mice were perfused with phosphate-buffered saline (PBS) containing 5 U/mL heparin, followed by a second perfusion with PBS. Hearts were obtained, atria were removed, and ventricles were weighed. The upper half of hearts was stored in paraformaldehyde and the apex was cut in small pieces, frozen in liquid nitrogen, and stored at −80°C. Tibia length was measured for normalization of ventricular weights. 25 Blood was collected in Microvette® tubes (Sarstedt, Germany) stored on ice for 30 minutes to induce coagulation and centrifuged at 4°C with 17,000×g for 25 minutes. Serum was used for the determination of TH levels.
Gene expression analysis
Total RNA was isolated from 20–30 mg heart tissue (QIAshredder and RNeasy Mini Kit, Qiagen) and eluted in 20 µL of RNase-free water. Total RNA concentration was measured with a NanoDrop2000. RNA integrity was tested on a denaturating 1.2% agarose gel (RNA-free water). Samples were incubated at 65°C for 10 minutes before being loaded on the agarose gel and visualized at a Molecular Imager® VersaDoc™. In total, 1 µg of total RNA was transcribed into cDNA by using a SuperScript™ III Reverse Transcriptase kit (Invitrogen) and random hexamer primers. Gene expression was normalized to that of three reference genes (18S, Gapdh, and Pol2a2). For analysis and calculation of gene expression, only Ct values below 35 cycles were evaluated using the efficiency corrected method. 26 Primer sequences are listed in Supplementary Table S1.
TH serum concentration
TH serum concentrations were measured with commercial FT3, FT4, and TT4 ELISA kits (DRG Instruments GmbH, Marburg) on a VersaMax Microplate Reader (Molecular Devices, Biberach). Minimum detectable levels of TH were 0.5 μg/dL for TT4, 0.05 ng/dL for FT4, and 0.05 pg/mL for FT3.
Statistics and software
Data were analyzed with GraphPad Prism 6 (GraphPad, San Diego, USA) and, if not otherwise noted, presented as mean ± standard error of mean (SEM). To test for significance, we used one-way ANOVA with Tukey’s multiple comparison test for comparison of all groups, Sidak’s multiple comparison test for comparison of selected groups, and Dunnett’s multiple comparison test for comparing all groups to one reference group. Two-way ANOVA with Bonferroni’s post hoc correction was used for echocardiography evaluation. Multiple t test was used to compare hourly averaged heart rate values measured over three days. Differences were considered significant with p < 0.05.
Results
T3 treatment induces ventricular growth via noncanonical TRα signaling
We compared posterior and interventricular wall diameters (LVPW;d and IVS;d) of male TRαKO, TRαGS, and TRβKO mice with their respective WT littermates with echocardiography before and after 7 weeks of T3 treatment (400 ng/mL), starting at an age of 8–10 weeks. T3 treatment for 7 weeks led to 1.5–2.0-fold increased FT3 serum concentration in all genotypes with FT4 concentrations below detection limit and significantly reduced TT4 (Supplementary Fig. S1).
Already at baseline, TRαKO mice had smaller LVPWs and lower relative ventricular weights compared to their WT littermates, whereas hearts of TRβKO mice did not differ from WT mice (Fig. 2A–C). In WT mice, 7 weeks of T3 treatment led to an increase in LVPW;d and IVS;d (WT littermates of TRα and TRβ mutant strains) (Fig. 2A–B). In contrast, T3 treatment did not induce an increase in wall diameters in TRαKO mice and their LVPW;d and IVS;d dimensions remained smaller than those of WT mice, indicating that absence of TRα prevented cardiac hypertrophy. A similar increase in LVPW;d as in WT mice was found in TRβKO mice and the IVS;d and LVPW;d diameters were not different from their WT littermates, demonstrating that absence of TRβ had no major influence on T3-induced cardiac hypertrophy. Unlike in TRαKO mice, the cardiac wall diameters of TRαGS mice were increased by T3 treatment, although the hypertrophic response was less pronounced than in WT controls. These data indicate that T3/TRα-mediated cardiac hypertrophy did not depend on canonical TRα signaling and was rather mediated by noncanonical TRα action.
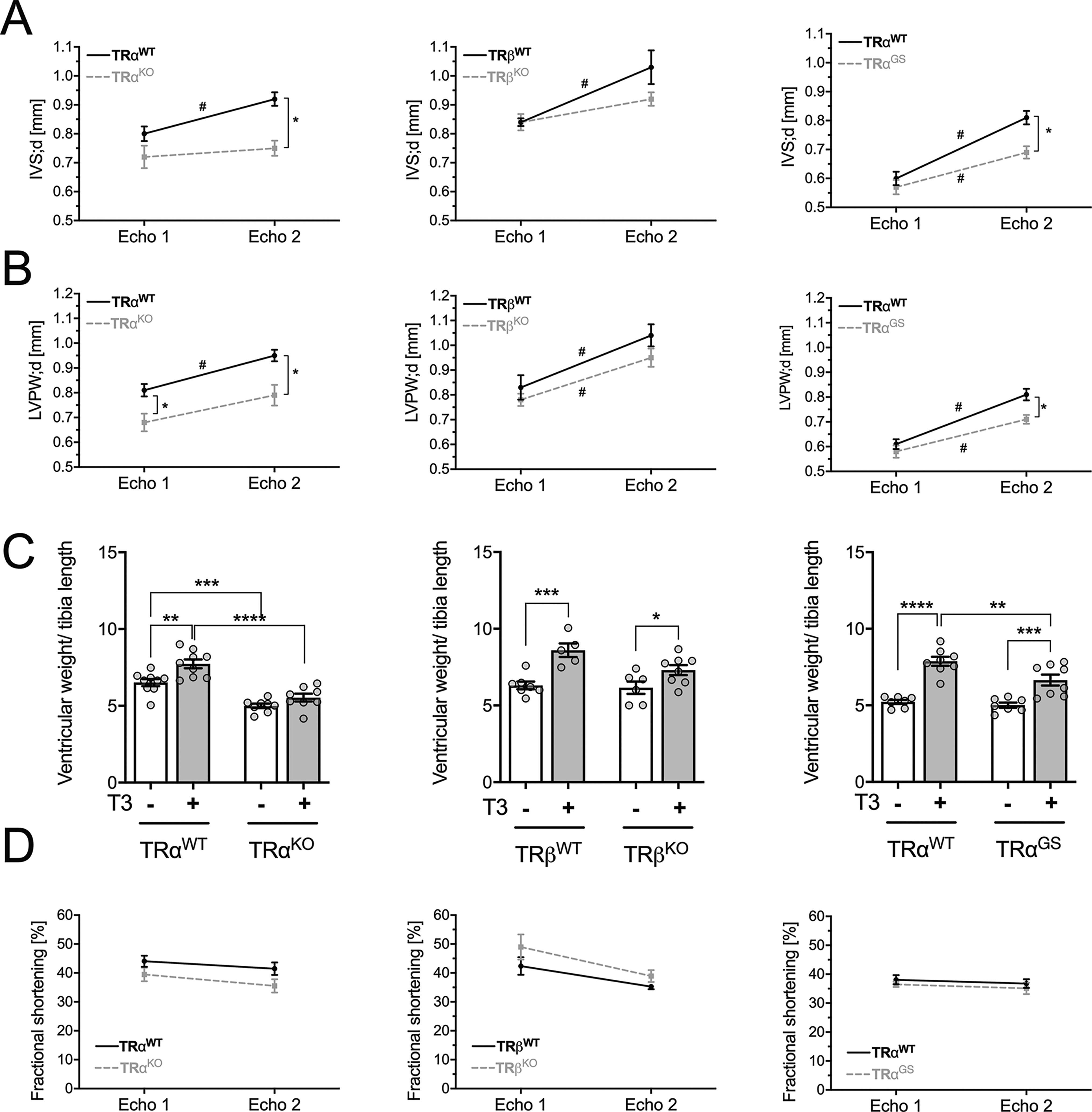
Echocardiography parameters: baseline and after T3 treatment of mice. Echocardiography measurements of male TRαWT, TRαKO, TRαGS, TRβWT, and TRβKO mice were conducted at experimental day 1 and after 7 weeks of T3 treatment.
At the end of the experiment, body weight, ventricular weight, and tibia length were measured (Supplementary Fig. S2). The ventricular weight/tibia length ratio (Fig. 2C) was increased by T3 treatment in all WT littermates compared to untreated WT mice. Ventricular weight/tibia length ratio was also increased in TRβKO mice but not in TRαKO mice (Fig. 2C), confirming that ventricular growth by T3 is mediated by TRα. Interestingly, the ventricular weight/tibia length ratio was also increased in TRαGS mice (Fig. 2C), further demonstrating that DNA binding of TRα was not required for this effect.
To assess cardiac function in response to 7 weeks of T3 treatment, fractional shortening was measured as a parameter for systolic function. Fractional shortening was not influenced by T3 in this experimental setting regardless of the genotype (Fig. 2D).
In accordance with increased echocardiographic wall diameters and ventricular weight/tibia length ratio, T3 treatment led to cardiomyocyte hypertrophy in WT and TRβKO mice, which was absent in TRαKO mice (Fig. 3). Again, the hypertrophic T3 effect was preserved in TRαGS mice, demonstrating that T3-induced cardiac growth is a consequence of cardiomyocyte hypertrophy and independent from TRα DNA binding.
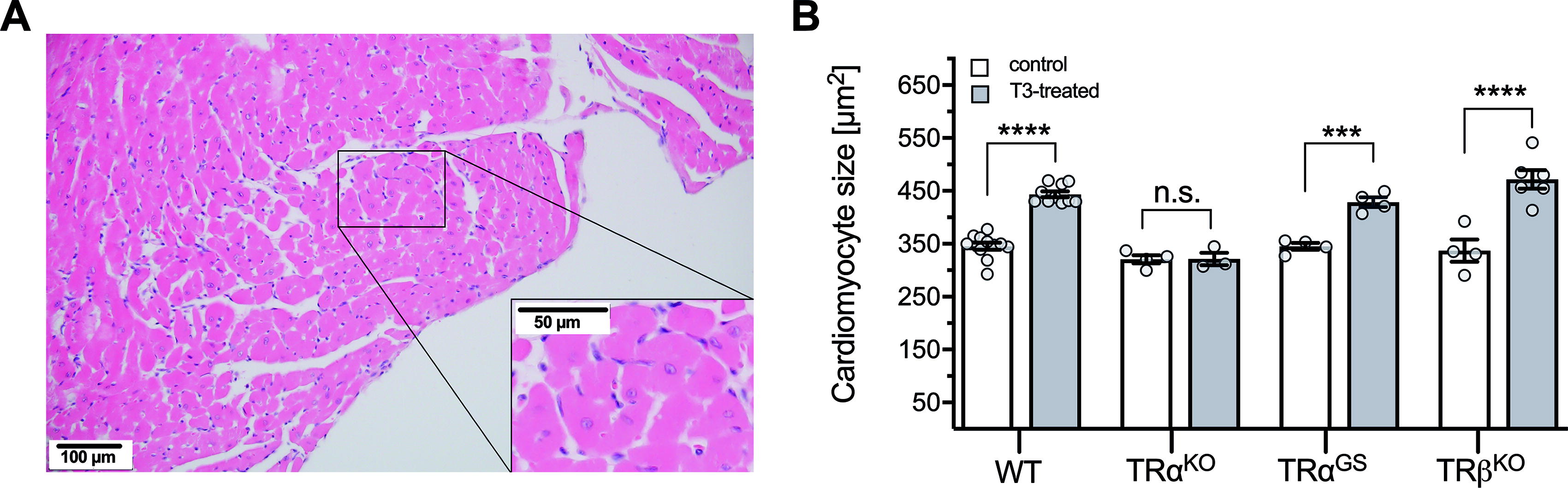
Cardiomyocyte size in untreated and T3-treated WT, TRαKO, TRαGS, and TRβKO hearts.
T3-mediated cardiac gene induction is regulated by TRα and TRβ
Next, we measured gene expression in control and T3-treated hearts from all genotypes. Thra and Thrb expression was absent or downregulated in respective TRKO hearts (Fig. 4). T3 treatment resulted in downregulation of Thra in WT and similarly in TRβKO and TRαGS hearts (Fig. 4). Expression of the positively regulated TH-target gene Myh6 differed only to a small degree between the genotypes and T3 treatment resulted only in a mild increase in TRαKO mice (Fig. 4). In contrast, the negatively regulated TH-target gene Myh7 was significantly reduced by T3 in all groups, which strongly suggests that T3-mediated repression of Myh7 can be mediated by both TRα and TRβ. Interestingly, loss of TRα or lack of DNA-binding ability of TRα in TRαKO or TRαGS mice, respectively, resulted in an increased basal expression of Myh7. This was not the case in TRβKO mice (Fig. 4). These data suggest that basal expression of Mhy7 is controlled by TRα rather than by TRβ, whereas T3-induced repression can be mediated by both TR isoforms, representing a complex isoform-specific regulation of Myh7.
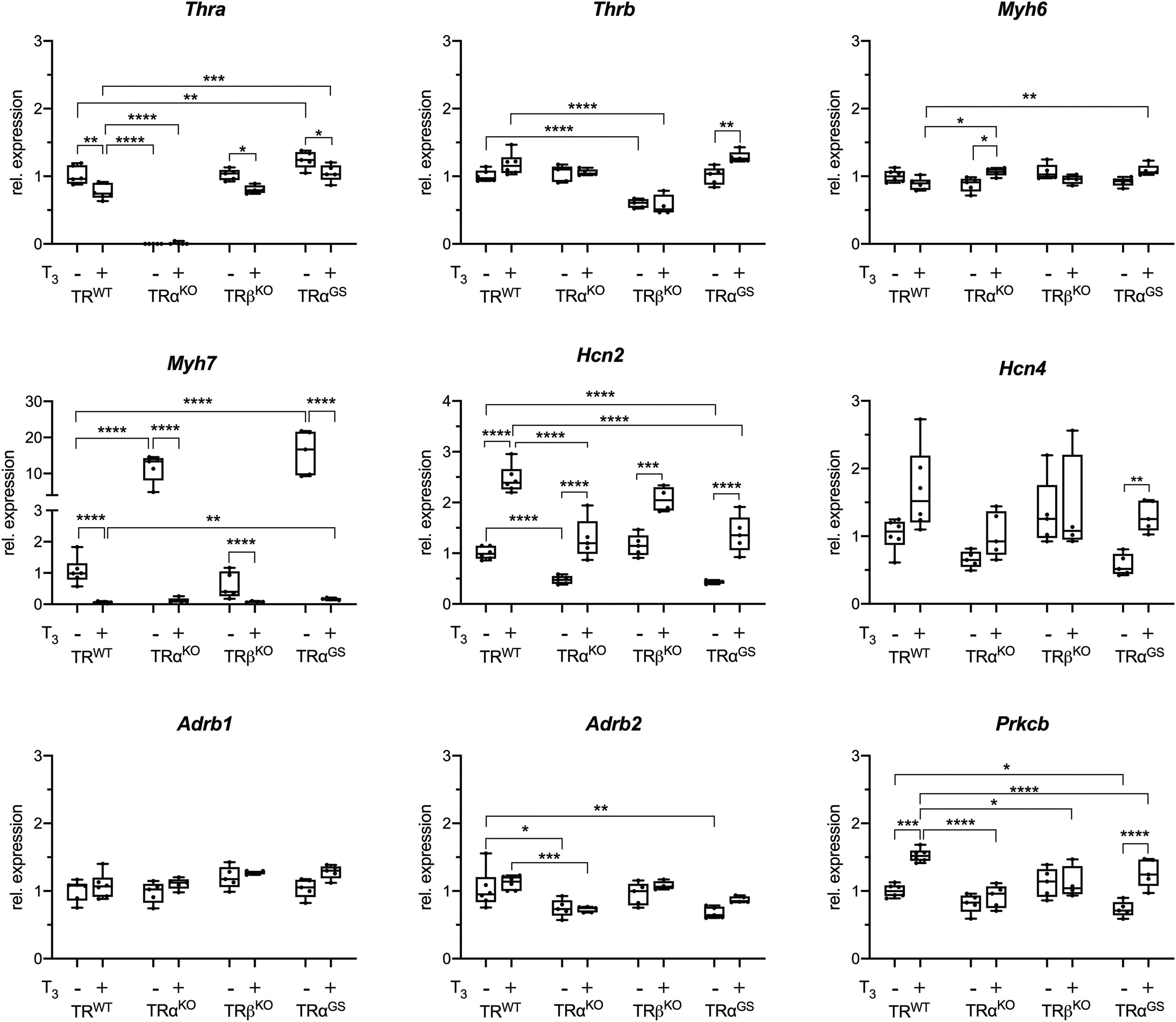
Relative gene expression in hearts of control and T3-treated mice. Relative gene expression of TH receptor α (Thra), TH receptor β (Thrb), myosin heavy chain 6 (Myh6), and 7 (Myh7), activated cyclic nucleotide gated potassium channel 2 (Hcn2) and 4 (Hcn4), beta-adrenergic receptor 1 (Adrb1) and 2 (Adrb2) as well as protein kinase C beta (Prkcb) (n = 5–6; one-way ANOVA with Sidak’s multiple comparison test [comparisons are male control vs. T3-treated and TRWT vs. TRαKO, TRβKO and TRαGS, respectively; values are shown as boxplots with whiskers [min to max]; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.).
Next, we measured the expression of T3-responsive pacemaker ion channels Hcn2 and Hcn4 that contribute to regulation of heart rate. Basal Hcn2 expression was reduced in TRαKO and TRαGS mice but not in TRβKO mice, suggesting that maintenance of basal expression of Hcn2 requires TRα but not TRβ. Interestingly, Hcn2 induction by T3 treatment was preserved in TRαKO, TRαGS, and TRβKO mice, which, similar to Myh7 repression, demonstrates that both, TRα and TRβ, can mediate the stimulatory TH effect. Hcn4 showed a similar expression pattern, although a higher variation within the groups resulted in less pronounced differences except for TRαGS, which still showed that DNA binding of TRα is not required for Hcn4 induction. Expression of Adrb1 and 2 was not responsive to T3 (Fig. 4).
Regarding a possible hypertrophic effect of TH treatment, we measured expression of protein kinase C beta (Prkcb), a kinase that is linked to cardiac hypertrophy. 27 –29 Basal expression of Prkcb was mildly reduced in TRαKO while reaching significance in TRαGS mice compared to WT mice, suggesting a TRα dependency. Interestingly, it seems to be the noncanonical mechanism that leads to Prkcb induction as Prkcb was induced by T3 in hearts of WT and TRαGS mice but not in TRαKO mice (Fig. 4).
Heart rate and its changes by TH serum concentrations are controlled by canonical TRα signaling
ECGs were recorded in conscious animals and cardiac electrical activity was detected noninvasively through the animals’ paws. Heart rate was reduced in TRαKO mice compared to WT controls (Fig. 5A). Heart rates of TRβKO and TRβGS mice, genotypes with intact TRα signaling, were not different from those of their respective WT littermates (Fig. 5A). Heart rate is therefore regulated by TRα, a classical, long-known physiological effect. 30
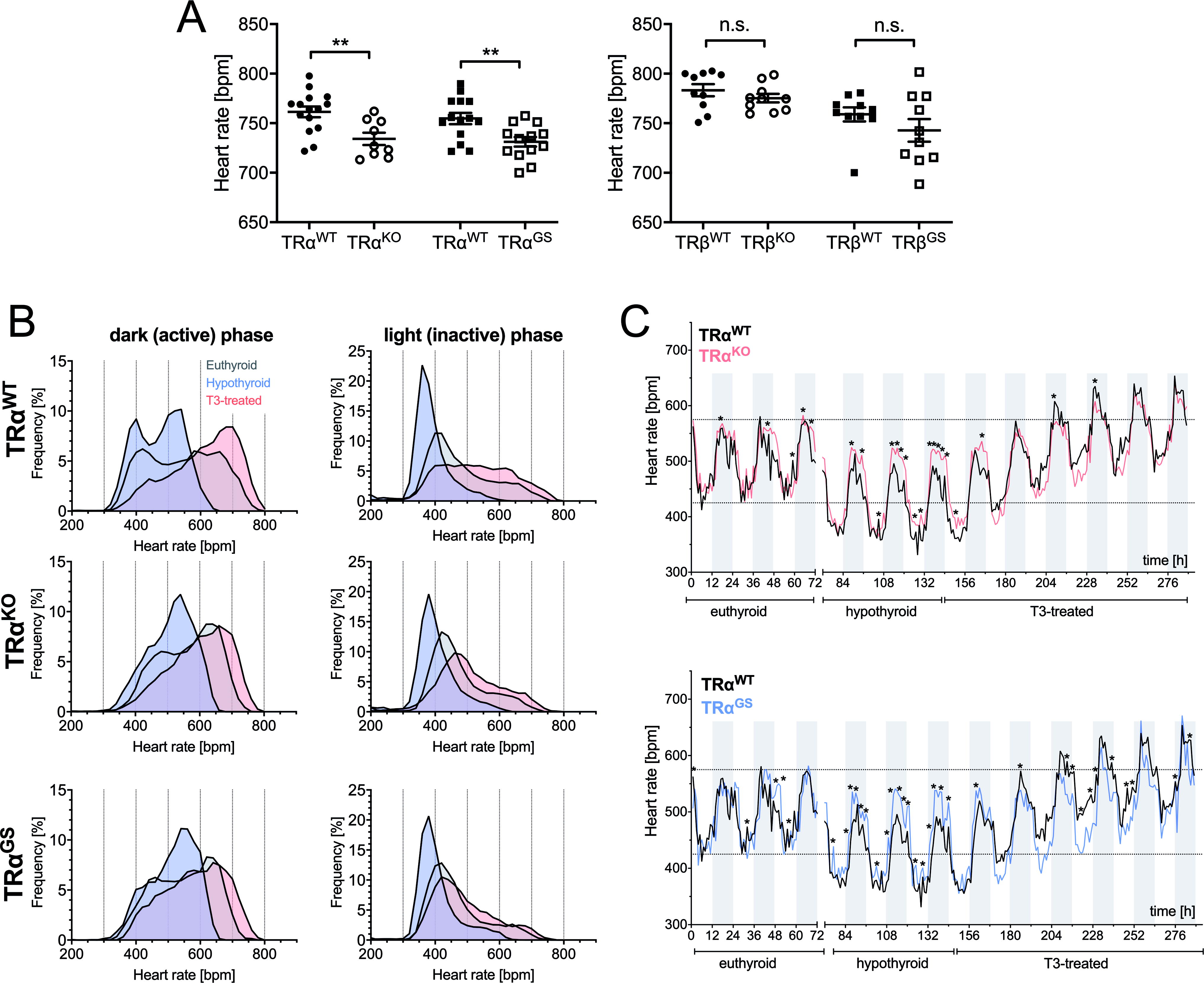
Heart rate is regulated by canonical TRα action with contribution from TRβ.
To analyze the response of heart rate in the TRα mutant mice to hypothyroidism and T3 treatment (successful T3 treatment was confirmed by ELISA measurements, Supplementary Fig. S3), we implanted telemetry devices into 4–5-month-old male mice for long-time monitoring of heart rates in conscious animals (WT, TRαKO, and TRαGS mice).
In WT mice, hypothyroidism resulted in lower heart rates and T3 treatment in higher heart rates (Fig. 5B). These changes in heart rate distribution, due to low or high TH, were more apparent in the active phase. Interestingly, the decrease and increase of heart rate in response to hypothyroidism or T3 treatment, respectively, was much less pronounced in TRαKO and TRαGS mice, especially in the active phase (Fig. 5B). During the inactive phase, there was only a mild adaptation due to hypothyroidism toward lower heart rates. Radiotelemetry recordings of TRαWT versus TRαKO and TRαWT versus TRαGS during the course of the protocol (untreated, methimazole/low iodine fed, and T3-treated) show that the deflection of heart rate was significantly reduced in TRαKO and TRαGS mice (Fig. 5C). These data demonstrate that the response of heart rate to the TH concentration, hypothyroidism or T3 treatment, is mainly regulated by canonical TRα signaling. Interestingly, heart rate was still modulated in TRαKO mice (Fig. 5B, C), demonstrating that TRβ contributes to heart rate regulation by TH.
Discussion
Main results from phenotyping of untreated and T3-treated WT, TRαKO, TRαGS, and TRβKO mice were that cardiac hypertrophy could be attributed to noncanonical TRα action, whereas heart rate regulation was mediated by canonical TRα action. As TH still modulated gene expression and heart rate in TRαKO mice, TRβ contributed to or even partially substituted for canonical TRα action.
Cardiac hypertrophy is a noncanonical TRα effect
TH induced cardiac hypertrophy in patients with untreated hyperthyroidism and in rodent models. 13,14,18,31 –34 While some mouse studies attributed this effect to TRβ action, there was also evidence of PI3K signaling pathway activation by TH, although this effect was not directly attributed to TRα. 17 –19 The present comparison of TR mutant mice showed that the hypertrophic effect of TH is predominantly mediated by TRα action and independent from DNA binding. These in vivo results are in line with the in vitro observation that an increased total protein content in cardiomyocytes, an index for overall hypertrophy, correlated with TRα1 signaling and not with TRβ. 15 Furthermore, TRα1-dependent cardiomyocyte hypertrophy involved p38 MAPK activation, which supports our conclusion that noncanonical signaling of TRα is relevant and provides a potential downstream target of noncanonical TRα action. 16
Already at baseline, TRαKO mice had smaller posterior and interventricular wall diameters (LVPW;d and IVS;d) and lower absolute and relative ventricular weights compared to their WT littermates, whereas hearts of TRβKO mice did not differ from WT mice. T3 treatment induced ventricular growth with increased wall diameters and ventricular weights in WT and TRβKO mice, but not in TRαKO mice. T3-induced cardiac hypertrophy was preserved in TRβKO mice, whereas absence of TRα completely abolished the T3 effect. Thus, the hypertrophic effect of T3 was mediated by TRα.
In contrast to TRαKO mice, TRαGS hearts were not smaller at baseline. Wall diameters, heart weight, and cardiomyocyte size of TRαGS mice increased significantly with T3 treatment. These results demonstrate that DNA binding is not required for TRα to mediate the hypertrophic effect of TH. One of the best characterized noncanonical effects of TRα is PI3K activation. 10,11,35 PI3K signaling is crucial in postnatal cardiac growth and constitutively active PI3K leads to cardiac hypertrophy with increased cardiomyocyte size, 36 similar to T3-treated WT and TRαGS mice, further supporting the concept that noncanonical DNA-binding independent TRα action underlies T3-induced cardiac growth.
We did not observe a change in cardiac function after 7 weeks of T3 treatment. This is not necessarily surprising, because even in mice with constitutively active PI3K fractional shortening was not changed. 36 We studied T3 effects only on healthy hearts and hypothesize that effects of noncanonical T3 signaling may become apparent in heart failure.
A more general question is whether TH-induced cardiac hypertrophy is a response to systemic activation of metabolism with increased demand of blood flow, a consequence of central activation of the sympathetic nervous system by TH or a direct heart intrinsic effect of TH. To determine precisely where T3 and TRα act would require studies in cell-type specific TR KO and KI mice, while we here studied global TR KO and KI mice. Therefore, our results do not allow to infer the site or cell-type of TH action. As our data from TR mouse models demonstrate that noncanonical TRα signaling is mainly responsible for cardiac hypertrophy and PI3K activation in mouse hearts was necessary for cardiac hypertrophy, 17 –19 we hypothesize that cardiac hypertrophy is a heart intrinsic effect of TH.
Absence of TRα can be compensated by TRβ
Myh7 encodes for a myosin heavy chain beta (MHC-β) isoform (slow twitch) expressed primarily in the heart and is repressed by TH. In contrast to untreated WT mice, basal Myh7 expression was not repressed in TRαKO mice and TRαGS mice, mice without TRα DNA binding, which demonstrates that TRα and its canonical mode of action mediate Mhy7 repression. T3 treatment repressed Mhy7 expression in WT mice. Strikingly, Mhy7 was as completely repressed in T3-treated TRαKO and TRαGS mice as in WT mice. This response to T3 must be independent from TRα. Our interpretation is that in the absence of TRα, TRβ substitutes for TRα and leads to Myh7 repression. The pattern of Hcn2 expression supports this hypothesis further: Hcn2 was induced by T3 not only in WT mice but also in TRαKO and TRαGS mice, which again must be independent from TRα, suggestive of substitution by TRβ. In TRβKO mice, T3 induced Hcn2 and repressed Mhy7, which suggests that to a certain degree both TR isoforms can substitute for each other’s canonical action. This may be the case for many genes and several organs because an in vitro study in neural C17.2 cells found that a substantial fraction, but not all, of the T3 target genes display a preference for one of the two receptor isoforms. 37 More than one-third of TR binding sites were shared by both TR isoforms, suggesting that both receptors contribute to regulation of the same genes as we found here in vivo. Prkcb showed an interesting expression pattern with T3-induced expression in WT and TRαGS but not TRαKO mice. Its expression seems to be independent from of TRα DNA-binding, a pattern we had previously observed for TRβGS-repressed genes in liver (Scd1, Fasn), 7 suggesting that noncanonical TRα action regulates gene expression although the mechanism remains to be determined. However, other than would be expected, Prkcb was not induced by T3 in TRβKO mice despite the present TRα. This is a puzzling observation for which we have no immediate explanation. Theoretically, a cooperation between TRβ and noncanonical TRα signaling in Prkcb induction would be compatible with the results, explaining an induction only when both are present (in WT and TRαGS mice), but not when one is missing (in TRαKO and TRβKO mice).
Heart rate
We had previously seen in an ex vivo model that heart rates of hyperthyroid TRαKO and TRαGS mouse hearts were comparable to WT mouse hearts. 38 Our interpretation was that residual TRβ expression may partially compensate for the absence of TRα. Here, in vivo heart rate measured with radio telemetry was again responsive to TH in global TRαKO and TRαGS mice. In both genotypes, heart rate was shifted in hypothyroidism and with T3 treatment, although with a narrower range than in WT mice. This regulation is most likely mediated through TRβ, apparently exerting the same function as the TRα, which is in line with previous observations. 39 While basal heart rate in ECG was clearly dependent on TRα, long-term comparison of hypothyroidism and T3 treatment with telemetry revealed a TH-mediated, but TRα-independent mechanism. Possibly, canonical TRα action controls basal and circadian heart rate while long-term adaption of heart rate in thyroid dysfunctions is likely to be controlled by TRβ.
Here, we studied male mice. As sex-specific differences in cardiac hypertrophy development and hypertrophic signaling have been reported, 40,41 it remains to be determined whether these results can be extended to female mice.
Conclusions
As gene expression (Myh7, Hcn2) and heart rate, both canonically regulated, were responsive to T3 treatment in TRαKO mice, we conclude that absence of canonical TRα signaling was compensated by TRβ. In contrast, T3 treatment for 7 weeks could not induce cardiac hypertrophy in TRαKO mice and TRβ could not substitute for this noncanonical TRα effect. We conclude that TRβ can substitute for canonical, but not noncanonical TRα signaling. This difference can be explained mechanistically. While TRα and TRβ share a large part of DNA binding sites, explaining overlapping canonical signaling, their noncanonical signaling has evolved differently after emergence of the two TR isoforms. 42 For example, the SH2 binding motif in TRβ that is crucial for TRβ/p85 association and PI3K activation is not present in TRα. Consequently, noncanonical signaling seen for TRβ was absent with TRα in CHO and rat pituitary cells. 6 Hence, TRα and TRβ are unlikely to be able to substitute for each other’s noncanonical signaling and this concept is supported here with in vivo data.
The present phenotypic observations from TR mutant mice may have implications beyond cardiac gene expression and heart rate. Currently, differences in physiological effects of TRα and TRβ are attributed to TR isoform-specific target genes or the relative expression of TR isoforms in cells. Based on our current results, we suggest that the difference in noncanonical TR action between TRα and TRβ also contributes to phenotypic TRα and TRβ differences.
In conclusion, the comparison of physiological T3 effects in WT and TR male mutant mice demonstrates that ventricular growth is largely mediated by noncanonical TRα action, whereas cardiac gene induction and heart rate regulation are mainly regulated by canonical TRα action. TRβ can partially substitute for absent canonical but not noncanonical TRα action.
Footnotes
Acknowledgments
We thank Konstanze Schättel, Andrea Jaeger, Kathrin Strumann, Jonas Bodmann, and the staff of the IMCES facility for expert technical assistance. We are grateful for the continued dedicated support from Prof. Dr. G. Hilken, Dr. A. Wißmann, Dr. P. Dammann, and the staff of the core animal facility at the University Hospital Essen.
Authors’ Contributions
L.C.M. conceived the project. Experiments were designed by D.G., G.S.H., N.S., D.F., K.L., and L.C.M. D.G., G.S.H., S.C.G., J.P., E.T., S.D., and K.L. performed the experiments and D.G., G.S.H., S.C.G., J.P., D.S., P.S., E.T., S.D., N.S., H.F., V.G.-D., M.H.A., J.M., D.R.E., D.F., K.L., and L.C.M. analyzed and interpreted data. H.F., V.G.-D., and M.H.A. designed and supervised the heart phenotyping at the German Mouse Clinic. D.G., G.S.H., and L.C.M. wrote the manuscript and all authors contributed to the final version.
Authors Disclosure Statement
The authors declare that no conflict of interest exists.
Funding Information
J.M., D.R.E., D.F., K.L., and L.C.M. are funded by Deutsche Forschungsgemeinschaft (DFG) Project-ID 424957847-TRR 296 LOCOTACT. L.C.M. was supported by DFG grant MO1018/2-2 and an IFORES grant from the Faculty of Medicine, University of Duisburg-Essen. K.L was supported by the German Ministry of Research and Education (BMBF; BMBF-ERK-Casting), the Ministry for Innovation, Science and Research of the Federal State of North Rhine Westphalia, and the DFG (SFB1116). MHA was funded by the German Federal Ministry of Education and Research (Infrafrontier grant 01KX1012) and the German Center for Diabetes Research (DZD).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Table S1