
Abstract
Background:
Mitochondrial dysfunction in the thyroid due to defective mitophagy has been observed in lymphocytic thyroiditis (LT). However, the effect of impaired mitophagy on the pathogenesis of LT is not well understood. The aim of this study is to investigate the role of mitophagy dysregulation in the thyroid gland.
Methods:
We analyzed RNA sequencing data of human thyroid glands with/without LT from Genotype-Tissue Expression (GTEx; n = 653) and performed RNA sequencing in thyroid glands of phosphatase and tensin homolog-induced putative protein kinase 1 (Pink1) knock-out and wild-type mice. We evaluated the phenotypic and histopathologic characteristics of the human (n = 16) and mouse thyroids. Additionally, we assessed cell proliferation, reactive oxygen species (ROS) production, and cytokine secretion of human thyroid epithelial cells (HTori-3) treated with PINK1 siRNA or a mitophagy inhibitor.
Results:
We found that expression of PINK1, a key regulator of mitophagy, was compromised in human thyroids with LT. Thyroid glands of Pink1-deficient mice exhibited increased inflammatory responses and nodular hyperplasia. Furthermore, mitophagy defects led to the production of pro-inflammatory cytokines and ROS in thyroid cells, resulting in immune cell recruitment. Notably, these mitophagy defects upregulated both the RNA expression and protein secretion of amphiregulin (AREG), an epidermal growth factor receptor (EGFR) ligand, in thyroid cells, while decreasing the protein expression of cAMP response element-binding protein (CREB), a transcription factor that suppresses AREG transcription. Finally, we demonstrated that aberrant cell proliferation in thyroid cells, driven by mitophagy defects, was mitigated after treatment with cetuximab, an EGFR inhibitor.
Conclusions:
In this study, we observed that mitophagy defects in the thyroid not only intensify inflammation through the accumulation of ROS, cytokine production, and immune cell recruitment but also contribute to hyperplasia via the EGFR pathway, facilitated by increased secretion of AREG from thyroid cells.
Introduction
Mitochondrial dysfunction has been found to play a critical role in the pathogenesis of various diseases, including metabolic, cardiovascular, neurodegenerative, and neuromuscular diseases. 1 Mitophagy, a mitochondrial quality and quantity control mechanism whereby damaged or superfluous mitochondria are eliminated, is key to maintaining cellular homeostasis, and defects in mitophagy contribute to excessive oxidative stress. 2,3 The thyroid gland is an endocrine organ with a high energy demand, in which oxidative processes are essential for thyroid hormone synthesis. As reactive oxygen species (ROS) are required in the initial stages of thyroid hormone production—during iodide oxidation by thyroid peroxidase (TPO)—the thyroid is particularly susceptible to oxidative damage. 4,5 Therefore, mitochondrial quality control for regulating ROS accumulation is crucial in thyroid cells, and mitophagy defects can lead to thyroid diseases. 6
Chronic lymphocytic thyroiditis (LT), also known as Hashimoto’s thyroiditis, is the most common cause of hypothyroidism in iodine-sufficient areas of the world and affects up to 10% of the global population. 7 LT is characterized by the infiltration of inflammatory cells in the thyroid gland and the production of autoantibodies to thyroid-specific antigens such as TPO and thyroglobulin. 8,9 Although the clinical presentation of LT may vary, it typically manifests as a gradual loss of thyroid function and nodular goiter formation with disease progression. 10 –12 The etiology of LT is multifactorial, and the mechanisms underlying the induction of immune cell recruitment and goitrous thyroid enlargement are not fully understood.
There is evidence of mitophagy defects in thyroid cells with LT, with a marked increase in the abundance of dysfunctional mitochondria with a swollen appearance and irregular cristae, 13 –15 suppressed autophagosome levels, 16 and excessive oxidative stress and mitochondrial ROS (mtROS). 17 Moreover, oncocytic change, characterized by cellular enlargement with abundant oxyphilic granular cytoplasm due to the accumulation of abnormal mitochondria, in thyroid follicular epithelial cells is a characteristic feature of LT. 18 However, the effect of impaired mitochondrial clearance on LT pathogenesis has not been elucidated.
In this study, we investigated the role of mitophagy defects in the thyroid, which might be involved in LT pathogenesis and progression, using transcriptomic and experimental approaches. Our findings may aid in the development of a promising new therapeutic strategy for protection against the advancement of LT.
Materials and Methods
Human thyroid RNA sequencing data and patient thyroid samples
The human thyroid mRNA expression and clinical phenotype annotation data were downloaded from the Genotype-Tissue Expression (GTEx) v8 (https://www.gtexportal.org/) and were used to compare the thyroid with or without LT (n = 653). Moreover, we included thyroid tissue samples from patients with both LT and nodular hyperplasia, as well as from patients without LT, for immunohistochemistry (IHC) analysis (n = 16). The clinical information of these patients is shown in Supplementary Table S1. All patients provided written informed consent, and this research was approved by the Institutional Review Board of CHA Bundang Medical Center (No. 2022-03-061).
Mice
Phosphatase and tensin homolog-induced putative protein kinase 1 (Pink1) knock-out (KO) mice were donated by Dr. Xiaoxi Zhuang (Department of Neurobiology, The University of Chicago, Chicago, IL, USA). The mice were backcrossed onto the C57BL/6 background for 20 generations. Pink1 wild-type (WT) mice served as controls. The animal procedures were approved by the CHA University Animal Care and Use Committee (No. 200012).
mRNA sequencing and data analysis
Total RNA was extracted from dissected mouse thyroids and spleens (n = 10 and n = 12, respectively) using NucleoZOL (Macherey Nagel, Düren, Germany) following the manufacturer’s instructions. An RNA library was constructed using the TruSeq Stranded mRNA LT Sample Prep Kit and sequenced on an Illumina TruSeq. For differentially expressed gene (DEG) analysis, we used the DESeq2 R package. 19 DEGs were defined as those with a |log2 fold change| >1 and a false discovery rate <0.05 and were subsequently used for functional enrichment analysis. Detailed information on mRNA sequencing data processing and analyses is provided in the Supplementary Data S1.
Serum TSH and free T4 measurement, histological analysis, and Western blot
Mouse serum thyroid-stimulating hormone (TSH) and free thyroxine (T4) levels were measured using a TSH enzyme-linked immunosorbent assay (ELISA) kit (#MPTMAG-49K; EMD Millipore, Billerica, MA, USA) and a free T4 ELISA kit (#1225-300, Monobind Inc., Lake Forest, CA, USA) and assessed using a Luminex 200 system (Thermo Fisher Scientific, Waltham, MA, USA).
IHC analysis for PINK1, 8-hydroxy-2’-deoxyguanosine (8-OHdG), and amphiregulin (AREG) was performed on formalin-fixed paraffin-embedded tissues of human thyroid glands with and without LT. The IHC score ranges from 0 to 9 and is calculated by multiplying the positive cell proportion score (0–3: 0 = 0%, 1 = 1%–25%, 2 = 25%–50%, 3 = 50%–100%) by the staining intensity score (0–3: 0 = no staining, 1 = weak staining, 2 = moderate staining, 3 = strong staining), as previously described. 20 IHC analysis for Ki67 and Cd45 was performed on mouse thyroid glands, and positive cells were quantified. ImageJ 21 and QuPath 22 software were used for the quantification.
Hematoxylin and eosin (H&E) staining, transmission electron microscopy, and Western blot analysis were performed on thyroid gland tissues of Pink1 KO and WT mice. Detailed methods are provided in the Supplementary Data S1.
Cells and in vitro studies
HTori-3, a normal human thyroid follicular cell line, and Jurkat, a human T-lymphocyte cell line, were used. HTori-3 cells were transfected with PINK1-siRNA or control-siRNA and were treated with mitochondrial division inhibitor 1 (Mdivi-1) (5 μM), cetuximab (50 μg/mL), transfection cell supernatant at 24 hours, or AREG (20 ng/mL). 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) and mitochondrial superoxide indicators (MitoSOX) (5 μM) staining were performed to quantify mitochondrial or cellular ROS production in HTori-3 cells and analyzed using a 570-nm excitation fluorescence microscope. Protein concentrations in supernatants of HTori-3 cells were determined using commercial human ELISA kits (R&D Systems, Minneapolis, MN, USA). mRNA levels were assessed by reverse transcription-quantitative polymerase chain reaction using FastStart Essential DNA Green Master (Roche, Basel, Switzerland), and cell proliferation was measured by the 3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (BIOSESANG, Sungnam, South Korea). For the transwell migration assay, conditioned medium (CM) from siPINK1- or siControl-transfected HTori-3 cells was placed in the lower chamber as a Jurkat cell chemoattractant, while Jurkat cells were seeded into Matrigel-coated upper transwell chambers and quantified after 24 hours of incubation. Detailed information on reagents and in vitro studies is provided in the Supplementary Data S1.
Statistics
Statistical analyses were conducted using two-sided Student’s t-test, Mann-Whitney U test, and Fisher’s exact test, depending on the data characteristics. Box plots or violin plots with box plots display the median and interquartile range (IQR), with whiskers extending to 1.5 times the IQR. The dashed line in the center of the plot indicates the median value of all data points. Bar graphs are presented as the mean ± SEM of multiple independent experiments. R version 4.2.1 (RRID:SCR_000432) was used for all statistical analyses. Statistical significance was defined as two-sided p-values <0.05.
Results
Downregulation of PINK1 was associated with LT and immune-related genes in the human thyroid
To investigate the changes in the expression of genes regulating mitophagy in LT, we used RNA sequencing data and histological information on human thyroid tissues (total, n = 653; no LT, n = 589; LT, n = 64) from the GTEx database. Of the top 10 mitophagy-related genes with the highest relevance score obtained from GeneCards, 23 PINK1 was the most significantly downregulated gene in thyroids with LT compared with those without LT (Fig. 1A, Supplementary Table S2). Subsequently, to further explore the biological role of PINK1 in the human thyroid, we divided the samples into low- and high-expression groups using the median value of PINK1 and obtained DEGs between the two groups. We then performed functional annotation analyses of the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways 24 and Gene Ontology (GO) terms 25 using the DEGs upregulated in the low-PINK1 group. All the top five KEGG pathways and most of the top five GO terms in each biological process and molecular function domain were found to be related to immune processes (Fig. 1B). Additionally, PINK1 expression was negatively correlated with the total immune cell infiltration score estimated using cell-type identification by estimating relative subsets of RNA transcripts x (CIBERSORTx), and similar trends were observed in 6 out of 22 subsets of immune cells, such as follicular helper T cells, CD8+ T cells, regulatory T cells, activated NK cells, naïve B cells, and M1 macrophages (Fig. 1C, Supplementary Fig. S1A), which are highly related to cytotoxicity and immune modulation. Furthermore, when analyzing the protein expression of PINK1 and 8-OHdG, an oxidative damage marker of mitochondrial DNA, using IHC in human thyroid tissues with and without LT (n = 8/each), we found that the cytoplasmic expression of PINK1 was lower (Fig. 1D) and that of 8-OHdG was higher in thyroid cells with LT (Fig. 1E). Additionally, the IHC scores for PINK1 and 8-OHdG were negatively correlated across the samples (Supplementary Fig. S1B). Taken together, PINK1 repression was observed in human thyroids with LT and was highly associated with mitochondrial oxidative damage and immune responses.
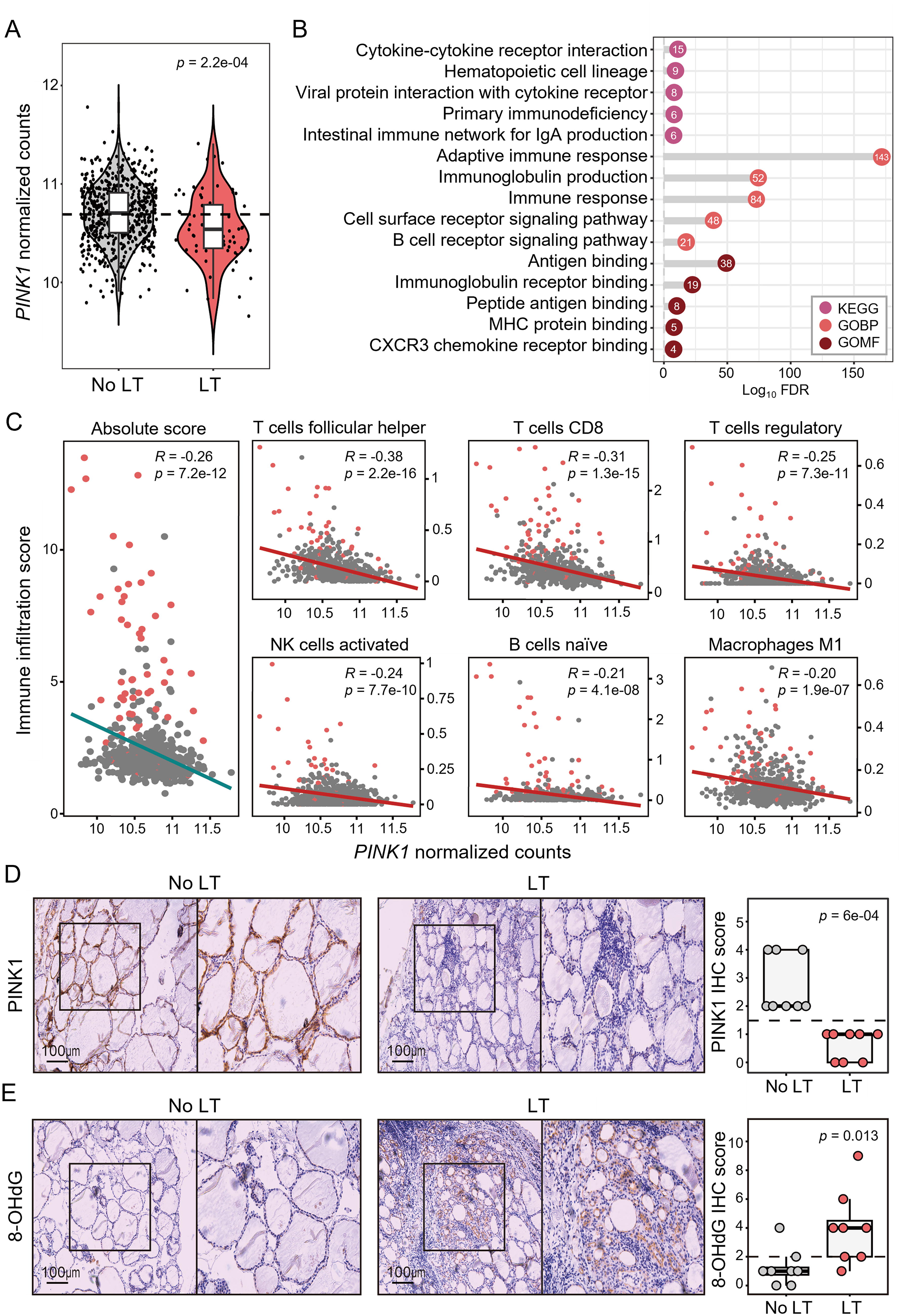
Downregulation of PINK1 was associated with lymphocytic thyroiditis (LT) and immune-related genes in human thyroids.
Mitophagy defects due to PINK1 loss induced thyroid hyperplasia and inflammation
To determine the effect of mitophagy defects on the thyroid, we used Pink1 KO and WT mice. Initially, we investigated histopathological alterations in the thyroid gland of Pink1 KO mice at 4, 12, and 24 months of age (Supplementary Fig. S2A and B). The thyroid tissues of 4-month-old mice exhibited no significant changes, while those of the 12-month-old mice demonstrated partial alterations. In contrast, the 24-month-old mice displayed pronounced and widespread changes in the thyroid gland. Consequently, we selected the 24-month-old Pink1 KO and WT mice for further analysis to investigate chronic changes (n = 8/each), akin to chronic LT.
The thyroid glands of Pink1 KO mice grossly showed goitrous and nodular changes (Fig. 2A), with a significant increase in thyroid gland weight (Fig. 2B). Moreover, serum TSH levels were elevated in Pink1 KO mice, while free T4 levels remained unchanged, indicating primary hypothyroidism in a subclinical state (Fig. 2C). However, there were no differences in the size or pathological findings of the anterior pituitary gland between the groups, regardless of Pink1 deletion (Supplementary Fig. S2C). Histological findings of thyroid glands showed that Pink1 WT mice maintained normal thyroid follicular cell structure (Fig. 2D). In contrast, Pink1 KO mice exhibited distinct histological findings of nodular hyperplasia, enlarged colloid-filled follicles, evident mononuclear immune cell infiltration, and lymphoid germinal center formation (Fig. 2D and 2E). Consistently, the percentages of Ki67-positive cells, a proliferation marker, and Cd45-positive cells, an immune cell marker, were significantly higher in Pink1 KO mice (Fig. 2F). Thyroid follicular cells with oncocytic changes due to aberrant accumulation of mitochondria were observed in the thyroids of Pink1 KO mice (Supplementary Fig. S3A and B). Furthermore, molecular markers of oncocytic changes such as Lmp2 expression and interferon gamma signaling 26,27 scores were significantly increased in Pink1 KO mice (Supplementary Fig. S3C). These histological and molecular findings were consistently observed in human thyroid tissues with LT (Supplementary Fig. S3D–F). Subsequently, we used transmission electron microscopy to analyze the ultrastructure of the mitochondria in thyroid cells. The thyroid cells of Pink1 WT mice showed normal mitochondria characterized by intact membrane, whereas those of Pink1 KO mice showed accumulation of abnormal mitochondria with swelling and bursting appearance (Fig. 2G). Therefore, long-term downregulation of Pink1 in the mouse thyroid resulted in aberrant cell proliferation, immune cell infiltration, and oncocytic changes with abnormal mitochondrial accumulation in thyroid cells, all of which are comparable to those seen in human chronic LT.
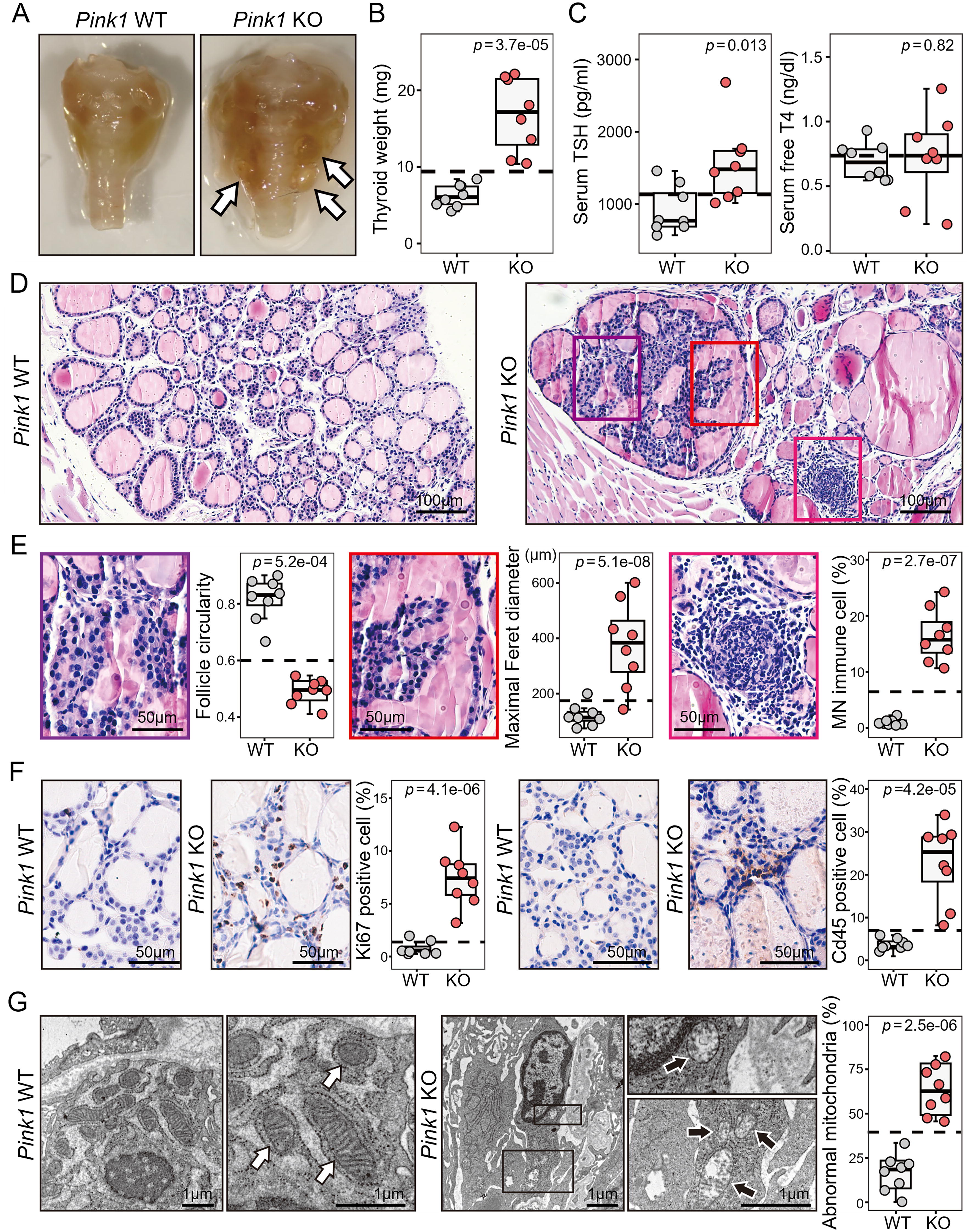
Mitophagy defects induced by Pink1-loss led to thyroid hyperplasia and inflammation in mouse thyroids.
Mitophagy defects due to PINK1 loss upregulated genes involved in immune response and damage-associated molecular patterns in the thyroid
To explore the molecular mechanisms of phenotypic changes in the thyroid due to PINK1-loss-induced mitophagy defects, RNA sequencing analysis was performed for the thyroid tissues of Pink1 KO and WT mice. We first confirmed that the thyroid glands of the Pink1 KO mice had a deletion of exons 4–7, as previously described 28 (Supplementary Fig. S4). Subsequently, we performed Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and Gene Ontology (GO) enrichment analyses with the DEGs between the thyroids of Pink1 KO and WT mice. Most pathways and GO terms enriched with upregulated DEGs in Pink1 KO mouse thyroids were associated with the activation of the immune response, which accounts for all of the top five KEGG pathways and eight of the top 10 GO terms (Fig. 3A). Moreover, the total immune infiltration score (Fig. 3B) and ROS-associated gene signature (Fig. 3C) were significantly higher in the thyroids of Pink1 KO mice than in those of Pink1 WT mice. As dysfunctional mitochondria have been reported to trigger sterile inflammatory reactions by activating damage-associated molecular pattern (DAMP) signaling pathways in several previous studies, 29,30 we hypothesized that DAMPs (endogenous molecules released in response to cellular damage that induce potent inflammatory responses as danger signals) can induce the activation of immune responses observed in PINK1-deficient thyroids. To test this hypothesis, we applied the scoring analysis to gene sets of biological pathways related to mitochondrial-derived DAMPs. All the DAMP-related gene set scores were significantly higher in Pink1 KO mouse thyroids than in Pink1 WT mouse thyroids, such as nucleotide-binding domain and leucine-rich repeat containing family pyrin domain containing protein 3 (NLRP3) inflammasome, toll-like receptor 9 (TLR9), stimulator of interferon genes (STING), and nuclear factor kappa light chain enhancer of activated B cells (NF-κB) (Fig. 3D). Consistent with the results of the RNA sequencing analysis in PINK1-deficient mouse thyroids, the ROS-related gene score and DAMP-related signaling pathway scores were significantly increased in human thyroids with LT (Supplementary Fig. S5A). Furthermore, to address the potential confounding factor that Pink1 deletion could be affecting immune cells, we performed RNA sequencing on spleens from Pink1 KO and WT mice (n = 6/each). Unlike the numerous DEGs observed in the thyroid, there were few DEGs between the two groups in the spleen (Supplementary Table S3), and therefore no enriched functional pathways were identified. In addition, pathways related to innate immunity and mitochondrial DAMPs showed no significant differences between the Pink1 KO and WT spleens (Supplementary Fig. S5B), suggesting that the impact of Pink1 deletion on immune cells themselves, as a confounding factor, is minimal.

Mitophagy defects exacerbated inflammation via upregulation of damage-associated molecular pattern (DAMP) signaling, mitochondrial reactive oxygen species (mtROS), and cytokine production in the thyroid.
Mitophagy defects exacerbated inflammation via cytokine production from thyroid cells as well as immune cell recruitment
A human thyroid cell line, HTori-3, was used to obtain insights into the mechanisms of DAMP signaling-related immune responses, and the cells were transfected with PINK1-siRNA or control-siRNA. PINK1 knockdown increased cellular and mtROS production as quantified using H2DCFDA and MitoSOX, respectively (Fig. 3E). The mRNA expression of DAMP signaling-related proinflammatory cytokines such as IL1B, IL6, and TNF in human thyroid cell lysates was upregulated in the presence of PINK1 knockdown (Supplementary Fig. S5C). The secreted protein levels of these cytokines in the CM from HTori-3 cells transfected with PINK1-siRNA (siPINK1-CM) were also higher than those in the CM from HTori-3 cells transfected with control-siRNA (siControl-CM) (Fig. 3F). Subsequently, we examined the effect of Mdivi-1, a mitophagy inhibitor that modulates mtROS production. 31 Consistent with the findings of PINK1 knockdown in HTori-3 cells, Mdivi-1-treated human thyroid cells showed increased expression and secretion of proinflammatory cytokines (IL1B, IL6, and TNF) (Supplementary Fig. S5D and Fig. 3G, respectively). Because immune cell infiltration was prominent in both mouse and human PINK1-deficient thyroid tissues, we determined whether thyroid cells with impaired mitophagy could stimulate immune cell chemotaxis. Interestingly, in the transwell migration assay, the migration potential of Jurkat cells, a human T lymphocyte line with wild-type PINK1, was significantly increased when treated with siPINK1-CM compared to siControl-CM (Supplementary Fig. S5E). Taken together, the results show that mitophagy defects in the thyroid induce inflammation by increasing the release of DAMP-related proinflammatory cytokines from the thyroid cell itself, which causes immune cell recruitment to the thyroid.
Mitophagy defects increased AREG expression and secretion in thyroid cells
Because mitophagy defects caused goitrous changes and hyperplasia of thyroid tissue in Pink1 KO mice, we investigated the mechanism of aberrant cell proliferation. In the GO functional annotations of RNA sequencing analysis of mouse thyroids, although most of the enriched gene sets were immune-related, a gene set of growth factor activity was significantly enriched with upregulated DEGs in Pink1 KO mice (Fig. 3A). Among the genes of this gene set, only amphiregulin (Areg in mice, AREG in humans), an epidermal growth factor receptor (EGFR) ligand, 32,33 satisfied the criteria for upregulated DEG in human thyroids with LT (Supplementary Table S4). To verify this, we first performed IHC staining of AREG in human thyroid tissues and demonstrated that thyroid follicular cells in thyroids with LT showed elevated AREG protein expression, whereas those in thyroids without LT did not (Fig. 4A). Additionally, it has been reported that the cAMP response element-binding protein (CREB) acts as a transcription repressor by binding to the cAMP response element sequences in the AREG promoter of thyroid cells; 34 moreover, CREB has also been shown to degrade under hypoxia. 35,36 Therefore, we investigated whether the molecular mechanism of increased AREG expression was associated with a decrease in its transcriptional repressor, CREB. We found that AREG expression was positively correlated with the hypoxia-related gene signature in mouse and human thyroid tissues (Supplementary Fig. S6A). Subsequently, we demonstrated that the protein expression of HIF1A, which is stabilized under hypoxic conditions, increased while CREB decreased in the thyroids of Pink1 KO mice compared to those of WT mice (Supplementary Fig. S6B and C). In vitro experiments showed that the mRNA expression of AREG was significantly increased by PINK1 knockdown and Mdivi-1 treatment in HTori-3 cells, as well as secreted protein levels of AREG in siPINK1-CM and Mdivi-1-treated-CM (Fig. 4B).
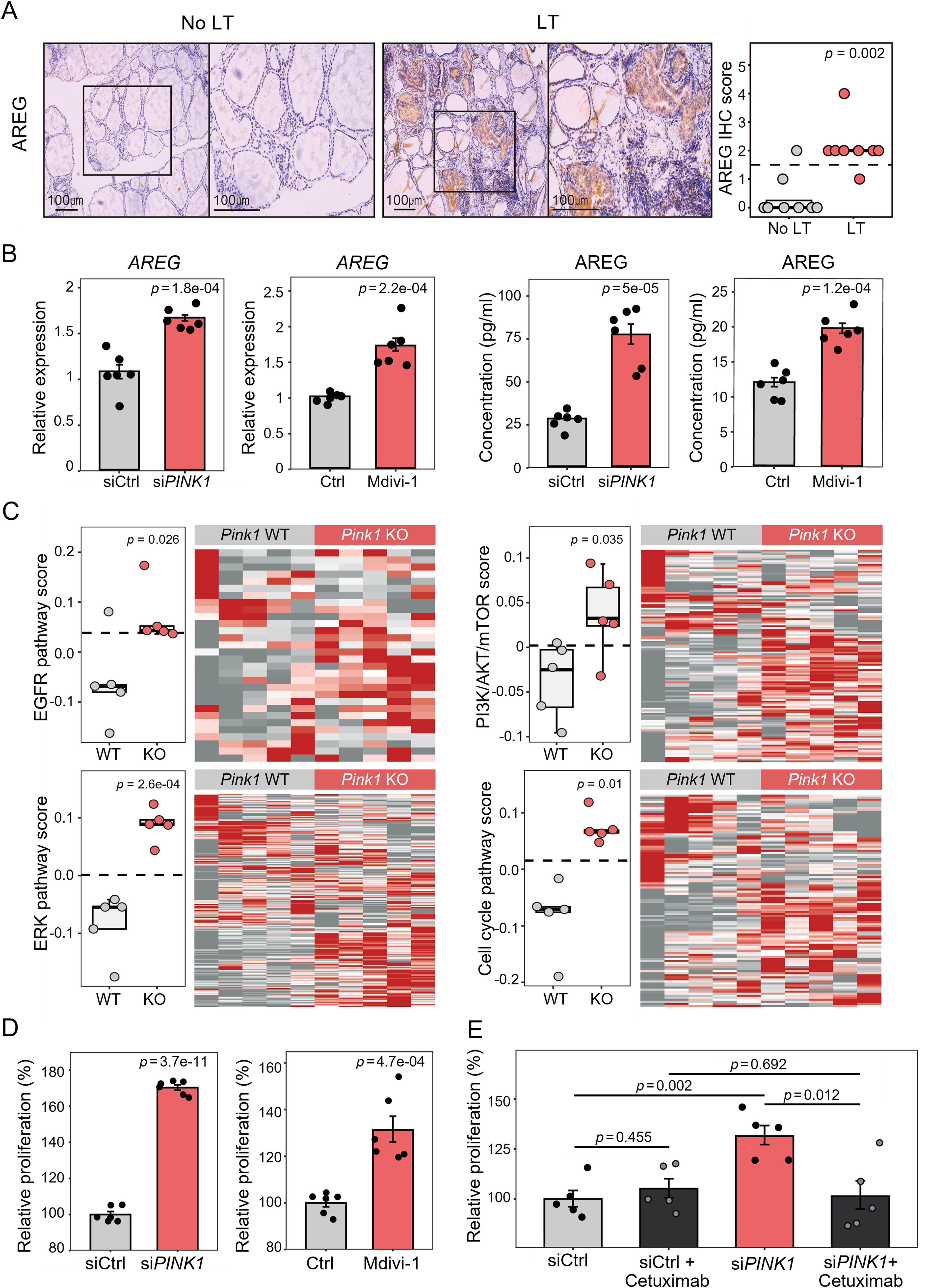
Mitophagy defects promoted aberrant cell proliferation by increasing amphiregulin (AREG) secretion and activating epidermal growth factor receptor (EGFR) pathway in the thyroid.
AREG promoted aberrant proliferation in mitophagy-deficient thyroid cells by activating EGFR signaling
We investigated whether increased AREG levels activate the EGFR pathway and lead to subsequent cell proliferation. Notably, the gene set scores of the EGFR pathway and its downstream signaling pathways, such as the RAS/RAF/MEK/ERK and PI3K/AKT/mTOR pathways, and the cell cycle-related gene set score were significantly increased in the thyroid glands of Pink1 KO mice (Fig. 4C). Consistent with the results of RNA sequencing analysis in PINK1-deficient mouse thyroids, the EGFR pathway-related gene set scores were significantly increased in human thyroids with LT (Supplementary Fig. S6D). Furthermore, the proliferation of HTori-3 human thyroid cells was significantly increased by PINK1 knockdown as well as by Mdivi-1 treatment (Fig. 4D). We also found that HTori-3 cell proliferation was promoted by treatment with siPINK1-CM as well as AREG itself (Supplementary Fig. S6E). Finally, to inhibit the effect of AREG-induced EGFR signaling activation, we treated HTori-3 cells transfected with PINK1-siRNA or control-siRNA with cetuximab, an EGFR inhibitor. 37 The increased proliferation of cells treated with PINK1-siRNA was reduced to a level similar to that of cells treated with control-siRNA by treatment with cetuximab (Fig. 4E). This suggested that EGFR inhibition effectively abrogated the stimulatory effect of impaired mitophagy on thyroid cell proliferation.
Discussion
Although many previous studies have shown that thyroid cells in LT contain accumulated abnormal mitochondria, it remains unclear as to how this is related to LT pathogenesis. 13 –15 In this study, we demonstrated that PINK1 downregulation in the human thyroid is associated with LT and immune-related genes. Using a Pink1 KO mouse model, we first reported the effect of mitophagy defects in the thyroid, which were found to induce inflammation and nodular hyperplasia. In vitro analyses of PINK1-deficient thyroid cells showed that inflammation is caused by increased ROS and cytokine production, as well as immune cell recruitment. Finally, mitophagy defects stimulated aberrant cell proliferation through AREG-induced EGFR signaling activation via CREB downregulation under hypoxia.
The PINK1-mediated mitophagy pathway is a key process for the selective elimination of damaged mitochondria, which show disruptive membrane potential. 38 Mitophagy is initiated by the accumulation of PINK1 following depolarization of damaged mitochondria. PINK1 promotes parkin recruitment and the subsequent ubiquitination of outer mitochondrial membrane proteins. The ubiquitinated mitochondria are engulfed by the autophagosome, which fuses with the lysosome and is subsequently degraded. Although mutations in PINK1 have been linked to early onset Parkinson’s disease, PINK1-mediated mitophagy has been studied in many organs to understand the mechanism and effect of aberrant mitochondrial dynamics. 28,39 –41 Since a genetic mitophagy defect has not been shown in human autoimmune thyroid disease, the use of the KO mouse model in this study may present a limitation. However, previous studies have reported mitophagy defects and oncocytic changes in thyroid cells associated with LT. 13 –15,18 Additionally, we demonstrated that PINK1 expression was significantly reduced in human thyroid tissues with LT compared to those without LT, at both the mRNA and protein levels.
Our findings showed that impaired mitophagy causes the accumulation of dysfunctional mitochondria in thyroid cells and triggers excessive mtROS production. Mori et al. 42 reported that in an iodide-augmented LT rat model, excessive iodide exposure leads to increased TNF alpha expression in thyroid follicular cells, with the proposed mechanism being increased mtROS. 43 mtROS oxidize mitochondrial DNA, which is recognized by the DAMP sensors NLRP3 and TLR9 signals. The activated DAMP signaling pathway upregulates the expression of proinflammatory cytokines, such as IL1B, IL6, and TNF, which trigger immune cell infiltration. 44 –48 Mitochondrial dysfunction due to defects in PINK1- and parkin-mediated mitophagy has been demonstrated to trigger inflammation via the STING pathway, 49 which is activated by DAMPs. A previous study reported that excess ROS in thyroid cells triggers proinflammatory cytokine secretion through the NF-κB-NLRP3 pathway activated by endogenous DAMPs. 50 Consistent with the results of previous studies, the thyroid of Pink1 KO mice showed significantly upregulated NLRP3, TLR9, STING, and NF-κB signaling pathway scores. Moreover, secretion of proinflammatory cytokines such as IL1B, IL6, and TNF was increased, and migration of human T cells was enhanced in siPINK1-transfected thyroid cells in vitro. Therefore, while autoimmunity remains the primary driver of LT, our findings suggest that impaired mitophagy-induced inflammation by excessive mtROS and DAMPs may be an additional stressor that exacerbates the autoimmune response.
Furthermore, we found abnormal cell proliferation in mitophagy-impaired thyroid tissues. Excessive mtROS are well-known hypoxia mediators 51,52 that stimulate hypoxia-induced transcription 51 and stabilize HIF1A. 53 –56 CREB, a transcriptional repressor of AREG, has been reported to decrease under hypoxia. 35,36 We showed increased HIF1A and decreased CREB protein expression in Pink1 KO mouse thyroids. We also demonstrated that AREG RNA expression and protein production increased in thyroid tissues and cells with defective mitophagy. Moreover, EGFR signaling pathways, including the PI3K-AKT-mTOR signaling pathway, the main downstream pathway, were activated by increased AREG levels. Activation of the mTOR pathway has been demonstrated to not only trigger cell growth but also regulate autophagy and mitophagy initiation. 57,58 We speculate that upregulation of the mTOR pathway via EGFR-EGFR ligand interaction suppresses the mitophagic machinery, resulting in a vicious cycle during the chronic disease course of LT, although further studies are needed to clarify this. Therefore, activation of the EGFR pathway by increased secretion of AREG from thyroid cells via hypoxia-induced decrease in CREB may be a potential mechanism of aberrant cell proliferation resulting in thyroid goiter in chronic LT. However, further rigorous molecular biological investigations are necessary to elucidate the precise mechanism by which mitophagy dysfunction leads to increased AREG secretion.
Most cases of LT evolve toward atrophy and hypothyroidism, but some exhibit goiter formation. The goitrous change in the thyroid can be stimulated by elevated TSH levels in chronic LT with hypothyroidism. 59 However, even if normal thyroid function is maintained, nodular goiter formation with chronic inflammation can develop as LT progresses. 60 This can cause cosmetic or obstructive symptoms that may require surgery. Our results showed that the blockage of activated EGFR signaling pathways normalized the aberrant proliferation of mitophagy-impaired thyroid cells. Therefore, targeting the EGFR pathway or AREG activity can be a therapeutic option for thyroid goitrous changes in LT for which there is no effective treatment other than thyroidectomy. Further preclinical studies are required to validate these results.
In conclusion, our study provides a mechanistic understanding of the role of PINK1-mediated mitophagy in LT progression. We discovered that a decrease in PINK1 expression leads to defective mitophagy in LT, resulting in increased mtROS. mtROS trigger DAMP-induced inflammation and cellular proliferation via hypoxia-mediated CREB degradation. Furthermore, our findings suggest that the autocrine activity of upregulated AREG, which turns on the EGFR pathway, is a putative driver of nodular goiter formation in LT (Fig. 5). Therefore, investigating strategies for promoting mitophagy may be a promising approach to eliminate damaged mitochondria and excessive mtROS while simultaneously protecting against LT progression. Additionally, blocking AREG or EGFR signaling may be a potential therapeutic target for nodular goitrous changes in LT.

Schematic diagram illustrating the effects of mitophagy defects in the thyroid. Mitophagy defects lead to the aberrant cell proliferation of thyroid follicular cells via the EGFR signaling cascade, mediated by AREG overexpression. In addition, it results in immune cell recruitment through the upregulation of mtROS, DAMPs, and the production of pro-inflammatory cytokines. Schema created with BioRender.com.
Footnotes
Acknowledgments
Availability of Data and Materials
Authors’ Contributions
H.S.L., S.-Y.L., and Y.S.S. designed this study. J.L., H.-J.A., M.-J.S., and J.-H.H. performed the experiments. H.S.L., J.L., and Y.S.S. collected and analyzed the data. H.S.L. and Y.S.S. contributed to article preparation. All authors proofread and approved the article.
Author Disclosure Statement
All other authors have no conflicts of interests to disclose.
Funding Information
This work was supported by the
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4