
Abstract
Background:
Diagnostic classification of thyroid malignancy is primarily accomplished through examination of histomorphological features and may be substantiated and clarified by molecular data. Individual molecular drivers show relatively robust and specific associations with histological subtypes of thyroid malignancy, including BRAF sequence variants and kinase gene fusions in papillary thyroid carcinoma, predominantly RAS variants in follicular-patterned neoplasia, and additional “late” mutations affecting TERT promoter, TP53, and the PI3K/AKT/PTEN pathway in high-grade malignancies. Given the oncogenic role of FGFR, particularly FGFR1-3, the goal of this study was to explore the role of FGFR in thyroid carcinoma biology.
Methods:
We completed a multicenter retrospective observational study for thyroid carcinomas with pathogenic alterations in the FGFR gene family. We performed this study by querying the molecular data accumulated for thyroid carcinomas from each center.
Results:
Overall, 5030 sequenced thyroid malignancies were reviewed, yielding 17 tumors with FGFR alterations, including 11 where FGFR was the primary molecular driver and 6 where FGFR was a secondary pathogenic alteration, with a subset for which there was available clinical follow-up data. Of the 11 carcinomas with an FGFR driver, 9 were gene fusions involving FGFR2:VCL (4 tumors), TG::FGFR1 (3 tumors), FGFR2::CIT, and FGFR2::SHTN1, and the remaining 2 were driven by FGFR1 amplification. In the 6 tumors where a canonical driver of thyroid neoplasia was present (5 cases) or no clear primary driver was detected (1 case), sequencing detected secondary FGFR2 p.W290C, p.Y375C, and p.N549K, as well as FGFR1 p.N546K in the respective tyrosine kinase domains, some at subclonal variant allele frequencies.
Conclusions:
This study presents the first description of a collection of thyroid carcinomas grouped by primary driver alterations in FGFR, as well as a cohort of thyroid tumors with secondary alterations that potentially lead to tumor progression or resistance to targeted therapy. Given the availability of small molecular inhibitors targeting oncogenic FGFR, this study emphasizes the significant implications for patients from identification of FGFR alterations as they are currently under-recognized in the literature and, most importantly, have potential novel treatment options.
Introduction
Most thyroid neoplasms originate from hormone-producing follicular epithelial cells. In the United States, thyroid cancer incidence has recently plateaued after decades of steady increases in case counts, attributed to improved screening and sensitivity of diagnostic testing. Papillary thyroid carcinoma (PTC) remains the most common endocrine malignancy, comprising up to 90% of primary thyroid carcinomas, follicular thyroid carcinoma (FTC) represents 5–15%, oncocytic thyroid carcinoma (OTC; formerly Hürthle cell) accounts for 3%, and anaplastic thyroid carcinoma (ATC) represents 1%. 1,2
Historically, the diagnosis of thyroid carcinoma subtypes has relied on histological features such as follicular versus papillary architecture, distinct nuclear and cytoplasmic features, encapsulation, and assessment for high-grade features represented by increased mitotic count and necrosis. These criteria and guidelines are outlined in the fifth edition (2022) of the WHO Classification of Endocrine and Neuroendocrine Tumors. 2 Over the past decade, the field has seen a major shift toward integration of tumor genotype and histological phenotype in clinical practice. 3 –7 Molecular analysis in the pre- and postoperative setting has become an essential component of patient management, including the necessity for or degree of surgery, and when neoadjuvant or postsurgical medical therapies may be beneficial. 8 –10
Of differentiated follicular cell-derived thyroid carcinomas, BRAF sequence variants followed by kinase gene fusions (e.g., RET, NTRK) are the main genetic drivers of most PTC subtypes, whereas RAS hotspots predominate driver alterations in follicular-patterned lesions driven by RAS-like molecular alterations, such as FTC. A landmark effort by The Cancer Genome Atlas (TCGA) project provided a detailed genomic, transcriptomic, and morphological association of PTC and follicular-patterned tumors, and highlighted differences between infiltrative BRAF-like PTC and invasive RAS-like tumors. 4 Genomic analyses of OTC have homoplasmic alterations of mitochondrial genes involved in the electron transport chain machinery, as well as near-complete haploidization of the nuclear genome as primary drivers, with a subset of near-haploid tumors showing subsequent genome duplication. 3,5,7 In more aggressive follicular cell-derived tumors, including differentiated high-grade thyroid carcinoma (DHGTC), poorly differentiated thyroid carcinoma (PDTC), and ATC, typical driver alterations of lower-grade thyroid tumors are frequently compounded by additional “late” alterations, including hotspot variants in the TERT promoter (most frequently hg19 chr5:g.1295228G>A; c.−124C>T; C228T, and hg19 chr5:g.1295250G>A; c.−146C>T; C250T), alterations in TP53, and mutations in the PI3K/AKT/PTEN pathway. 6,11 With few exceptions, these findings are consistent with multistep molecular progression toward dedifferentiation, aggressive tumor biology, and a poorer prognosis. 6,8,11 However, there are rare situations in which late molecular alterations, predominantly of the TERT promoter, are also present in more indolent lesions, 12 –14 raising questions of temporal and spatial drivers of varying biological potential.
The fibroblast growth factor (FGF) signaling pathway, mediated by the FGFR1-4 family of receptor tyrosine kinases, is important for cellular proliferation, prolonged survival, invasion, and differentiation. 15 Genetic alterations in FGFR are known mechanisms of oncogenesis and have been found most frequently in urothelial carcinoma among a variety of other tumors. 16 Following recent investigations demonstrating successful outcomes with FGFR-targeted therapy in solid tumors, including intrahepatic cholangiocarcinoma and other advanced tumors, 17 –20 we initiated a study to characterize genomic alterations of the FGFR gene family in thyroid tumors. Here, we present clinicopathological data from the first cohort of thyroid carcinomas with likely oncogenic FGFR1/2 primary driver alterations, an under-recognized molecular event in this setting. We also present clinicopathological data from patients with thyroid tumors harboring typical driver alterations and concomitant secondary FGFR1/2 variants, an important finding in the context of recent data demonstrating efficacious targeting of FGFR-mediated tumor resistance following RET-directed therapy. 21
Materials and Methods
Study design
To assess FGFR alterations within malignant thyroid tumors, we designed a multicenter retrospective observational study involving the Massachusetts General Hospital (MGH) Center for Integrated Diagnostics (CID), Foundation Medicine, Inc. (FMI), and Brigham and Women’s Hospital (BWH) Center for Advanced Molecular Diagnostics (CAMD). All molecular testing was performed as part of clinical care, with patients consented in accordance with each respective laboratory’s practices and policies. Clinicopathological data were collected where available, with missing data indicated per instance.
FGFR variant classification and case inclusion criteria
Sequencing archives were reviewed for thyroid tumors with FGFR alterations, including sequence variants (single nucleotide variants [SNV], insertions-deletions [indels]), gene fusions, and copy number variants [CNV]), and was collected and evaluated agnostically to reduce bias. Tumors without other known, typical drivers of thyroid neoplasia were considered positive for pathogenic FGFR primary driver alterations. Tumors either with a concurrent known driver of thyroid neoplasia (e.g., BRAF, RAS, DICER1, RET, NTRK1/3, large-scale chromosomal CNV) or with an FGFR SNV/indel at a subclonal variant allele frequency (VAF) were considered secondary molecular alterations. Secondary FGFR alterations were evaluated for pathogenicity and only those with reasonable evidence of “likely pathogenic” or “pathogenic” biological behavior in publicly available databases were included in the study.
Case identification and molecular analysis
Center for Integrated Diagnostics, Massachusetts General Hospital
This study was approved by the Mass General Brigham (MGB) Institutional Review Board (IRB; 2019P002183). We reviewed the results of DNA- and RNA-based targeted next generation sequencing (NGS) of thyroid tumors performed at the MGH CID between April 16, 2014 and June 30, 2023. Cases with SNV, indel, gene fusion, or CNV in FGFR1-4 were identified, and diagnostic slides were obtained and reviewed by 2 thyroid pathologists (PMS and ASF). Clinicopathologic information including patient demographics, surgical and molecular pathology findings, and clinical follow-up were obtained through the MGB electronic medical records (Epic Systems, Verona, WI, USA).
Targeted NGS was performed as part of routine clinical diagnostics using anchored multiplex-PCR-based 22 Archer NGS assays (Integrated DNA Technologies, Coralville, Iowa, USA) and the ion torrent-based Genexus assay (Thermo Fisher, Waltham, MA, USA). The target genes of these panels were selected for identification of diagnostic, prognostic, and therapeutically impactful alterations. Assays were performed in a Clinical Laboratory Improvement Amendments (CLIA)-accredited laboratory, analyzed by a clinically validated pipeline, and results were reviewed and interpreted by molecular pathologists. The Archer RNA-based assay targets FGFR (exon) fusion transcripts in FGFR1 (2, 8–10, 17), FGFR2 (2, 8–10, 17), and FGFR3 (8–10, 17, intron 17), whereas the DNA-based assay targets FGFR (exon) SNV/indels in FGFR1 (4, 7–8, 13, 15, 17), FGFR2 (7, 9, 12, 14), and FGFR3 (7–9, 14–16, 18), and CNV in FGFR1-3. The RNA-based Genexus assay targets intergenic fusions in FGFR1-3, whereas the DNA-based assay targets FGFR (codon) SNV/indels in FGFR1 (V592, K687), FGFR2 (S252, P253, K310, A314, A315, Y375, C382, M391, V395, H544, I547, N549, V564, E565, G613, L617, K641,A648, K659, V679), FGFR3 (R248, S249, G370, S371, Y373, G380, F384, A391, R399, V553, V555, D641, K650, V677, K715), and FGFR4 (V550), as well as CNV in FGFR1-3. A clinically validated fluorescence in situ hybridization (FISH) assay to assess for FGFR1 amplification was performed on one tumor (case 11). Cohort cases 1, 3, 7, 11, 14, and 17 were analyzed using the Archer RNA-based assay, cases 3 and 14 were tested on Genexus, and cases 7, 11, 14, and 17 were tested on the Archer DNA-based assay.
Foundation Medicine, Inc
The study was conducted under the Western Institutional Review Board approved protocol number 20152817. The FoundationCORE® research database at FMI was retrospectively queried for thyroid tumors sequenced using FoundationOne®CDx, which has been described previously. 23,24 This assay covers all coding exons of FGFR1-4 for SNV, indel, and CNV detection, and the test assesses select intronic regions for detection of FGFR1-3 rearrangements. Cohort cases 2, 4, 5, 6, 9, 10, 12, 13, 15, and 16 were analyzed on FoundationOne®CDx.
Center for Advanced Molecular Diagnostics, Brigham and Women’s Hospital
The inclusion of this case was approved by the MGB IRB (2016P001885). Cohort case 8 was tested at BWH CAMD on the OncoPanel targeted NGS assay, 25 targeting SNV, indels, gene rearrangements, and CNV.
Data visualization and statistical analysis
Gene fusion diagrams were generated using St. Jude’s PeCan. 26 Tables were generated using the gt package. 27 Summary statistics included mean or median, range, count, and percentage for a variety of demographic and clinicopathological factors. Images of read-level data from BAM files were acquired using the Integrative Genomics Viewer (Broad Institute, Cambridge, MA, USA).
Results
Overall cohort findings
In total, 5030 sequenced thyroid tumors were evaluated. Seventeen tumors met inclusion criteria (Fig. 1), all of which harbored alterations in FGFR1 (ENST00000425967) and FGFR2 (ENST00000457416). The identified cohort had a median age of 57 years (range: 24–87 years) and 59% females (Table 1). Ten tumors (59%) were primary, 5 (29%) recurrent, 1 tumor was from a metastasis (6%), and 1 the clinical context for one tumor was unavailable. Of 17 cases with FGFR alterations, FGFR1 and FGFR2 alterations identified in 11 carcinomas (65%) were considered primary driver alterations. Of the remaining 6 tumors (35%), 5 were found to have FGFR1 and FGFR2 alterations coinciding with known, canonical drivers of thyroid neoplasia, whereas case 16 had no clear primary driver and harbored a pathogenic FGFR2 variant at 1.1% VAF. No patients received prior FGFR inhibitor treatment.

Flow diagram of tumor selection for review in the study.
Summary of Patient Cohort of Thyroid Malignancies with FGFR Alterations (n = 17)
FGFR primary driver alterations in thyroid neoplasia
Among 11 tumors with FGFR1/2 primary driver alterations (Table 2), ranging in size from 1.0 to 7.4 cm, 6 tumors showed high-grade morphology, including 5 PDTC and 1 DHGTC arising in a PTC, and the remaining tumors included PTC subtypes. Molecular drivers in FGFR were predominantly gene fusions (9 tumors); two-thirds harbored 5’ FGFR2 with a recurrent exon 17 breakpoint (Fig. 2). FGFR2 gene fusion partners were predominantly VCL (4 cases, exon 2 breakpoint; Figs. 3 and 4), whereas other fusions harbored 3’ CIT (exon 23 breakpoint), and 3’ SHTN1 (exon 9 breakpoint). These cases had no concurrent sequence variants detected. Recurrent 3’ FGFR1 fusions were detected in 3 tumors with exon 3 (2 cases) and exon 10 (Fig. 5) breakpoints. FGFR1-translocated cases also had sequence variants identified by NGS, including TP53, PIK3CA, and TERT promoter C228T alterations, among others. The final driver alteration was FGFR1 amplification, found in two tumors, one identified by FISH in a tumor that also harbored a TERT promoter C228T alteration (Fig. 6). Multiple gene copy gains and losses were seen in this cohort, most notably a two-copy CDKN2A/CDKN2B deletion in case 5.
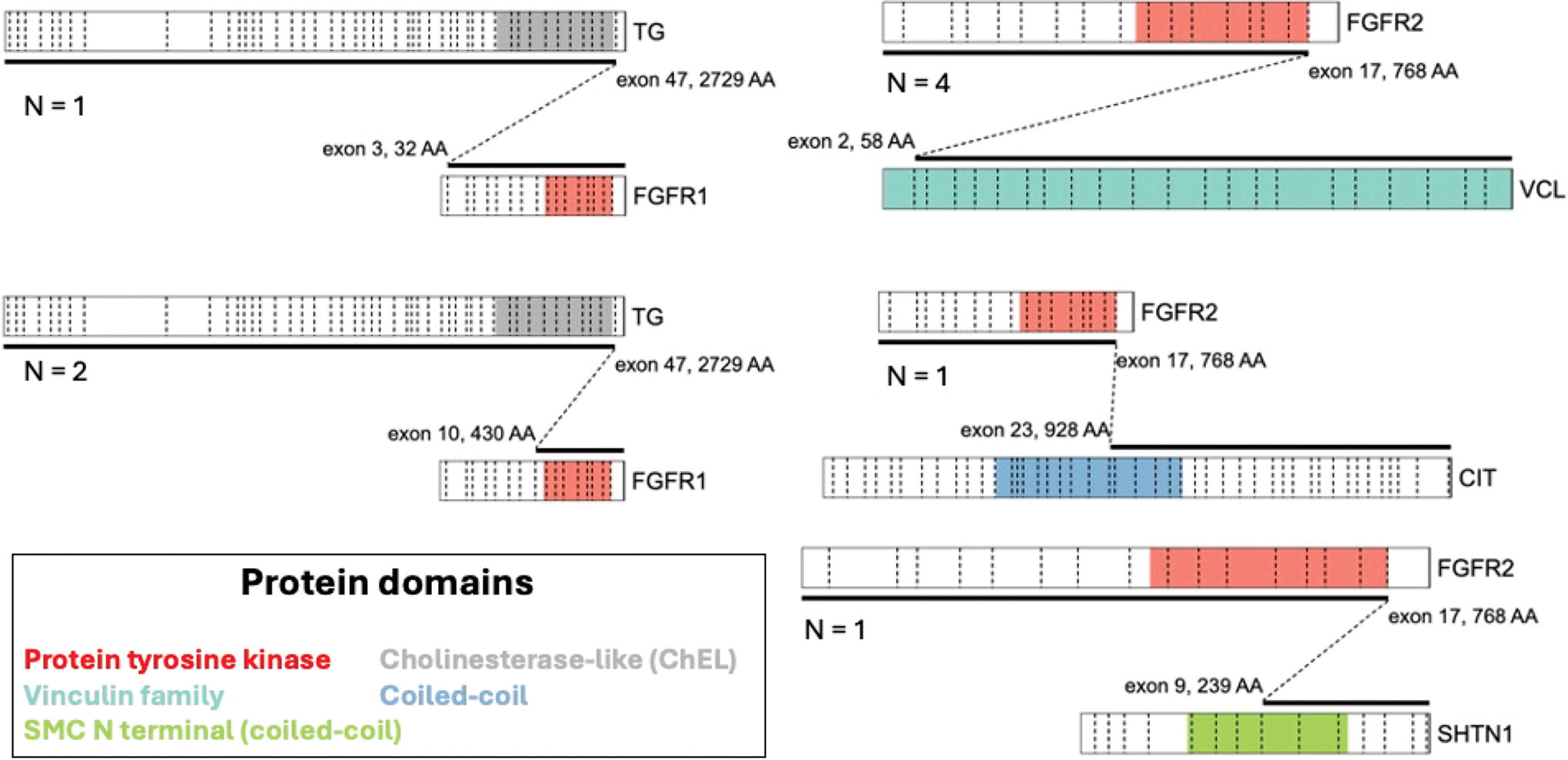
Schematics of the FGFR1 and FGFR2 gene fusions identified as primary driver alterations in the cohort. Fusions are depicted with 5’ fusion partner above 3’ fusion partner and with dashed diagonal lines indicating the exonic breakpoints. Dashed vertical lines delineate coding exons within each gene. Relevant protein domain annotations are color-coded. N, number of cases.
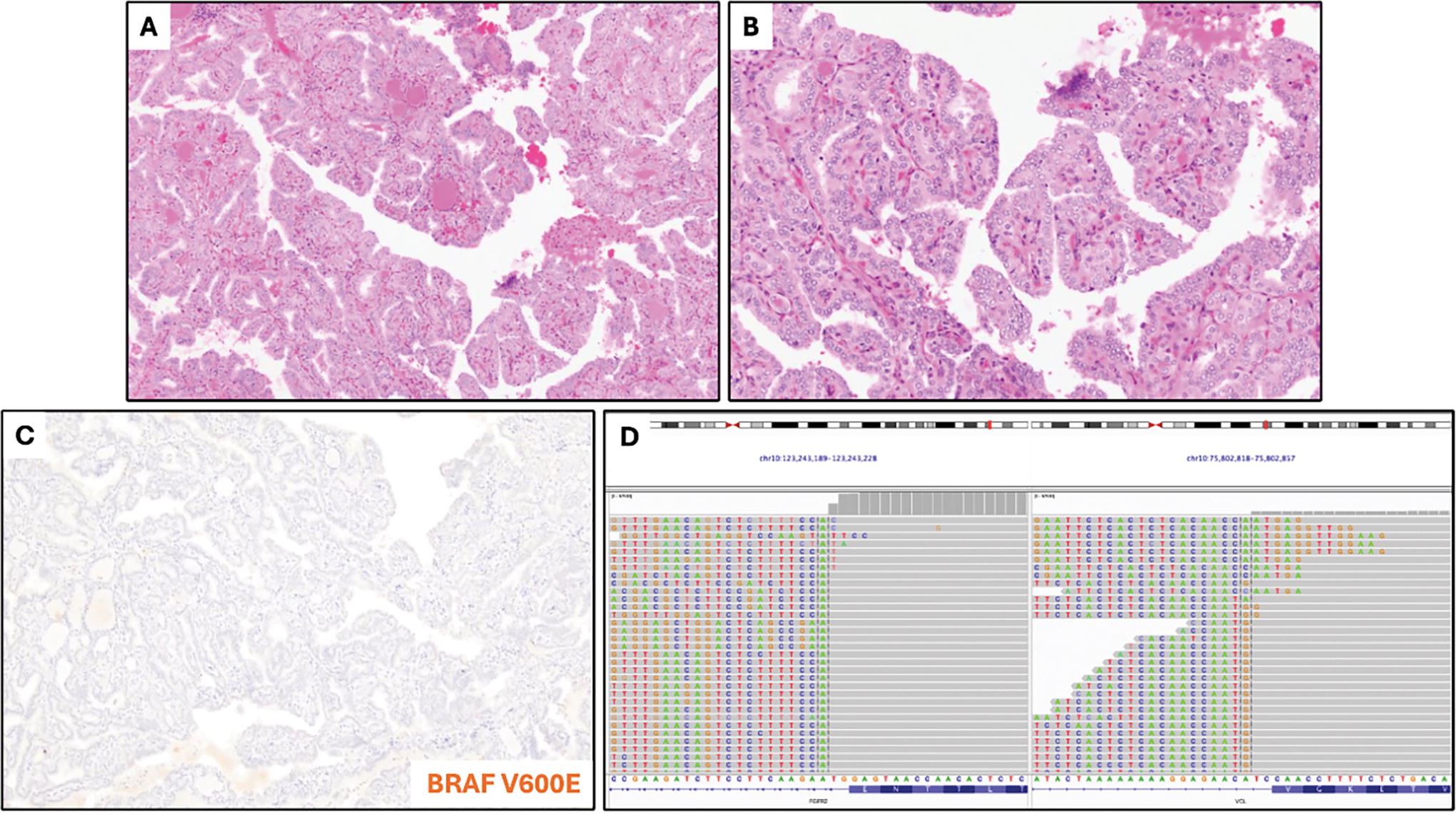
Case 1. Low-power
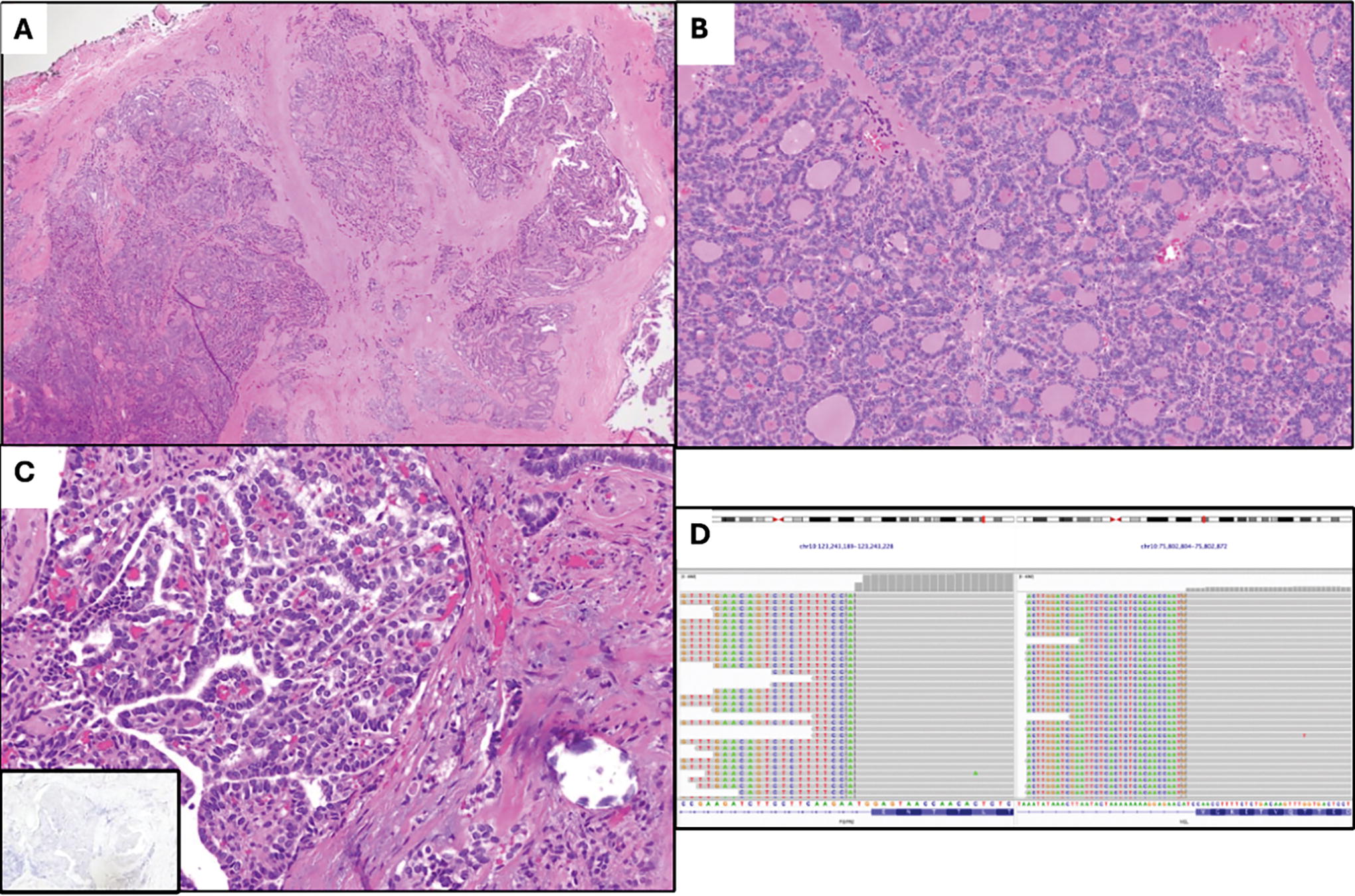
Case 3. Examination of this differentiated high-grade thyroid carcinoma arising in a papillary thyroid carcinoma at low magnification
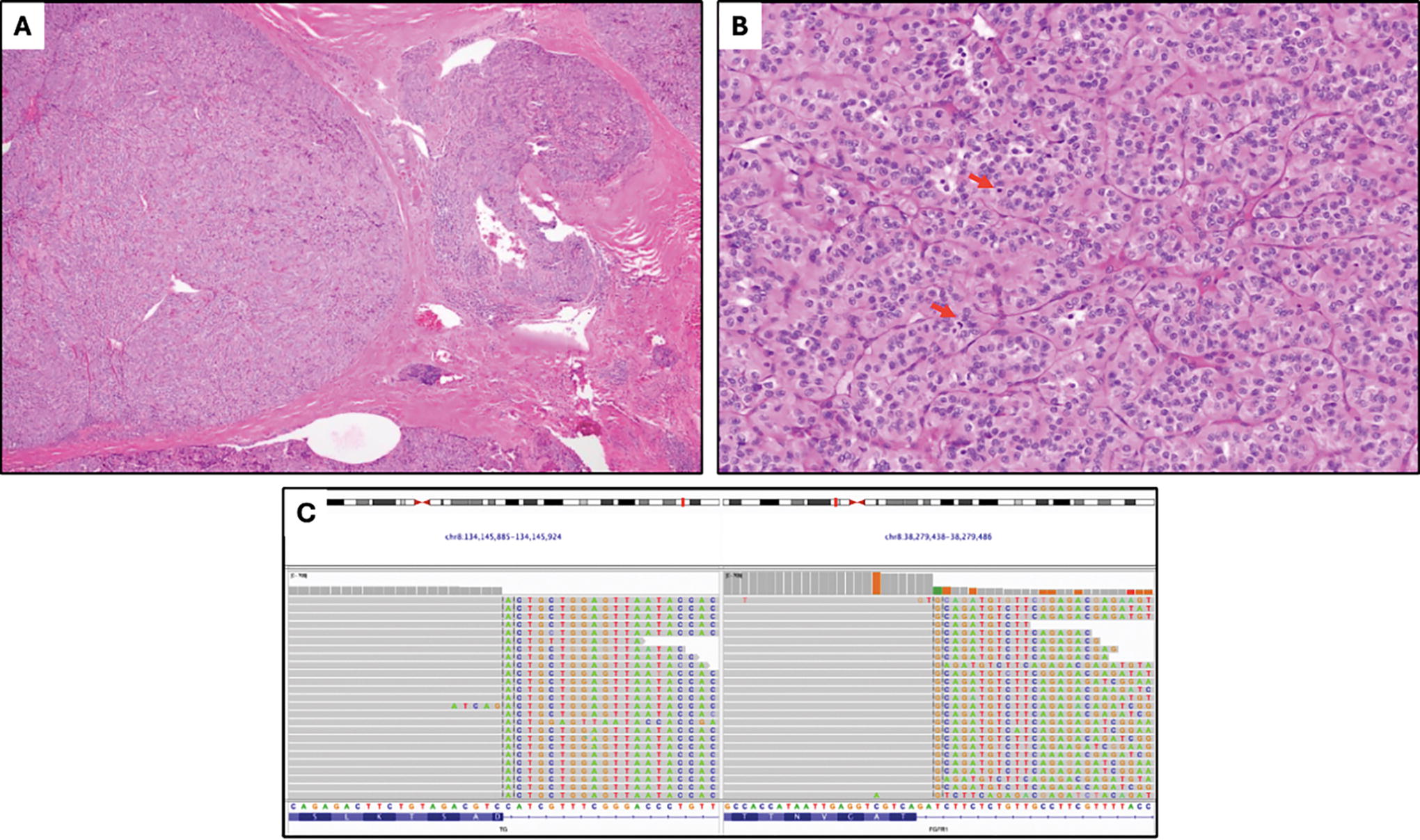
Case 7. A poorly differentiated thyroid carcinoma arising in widely invasive follicular thyroid carcinoma shows broad invasion that appears solid on low magnification
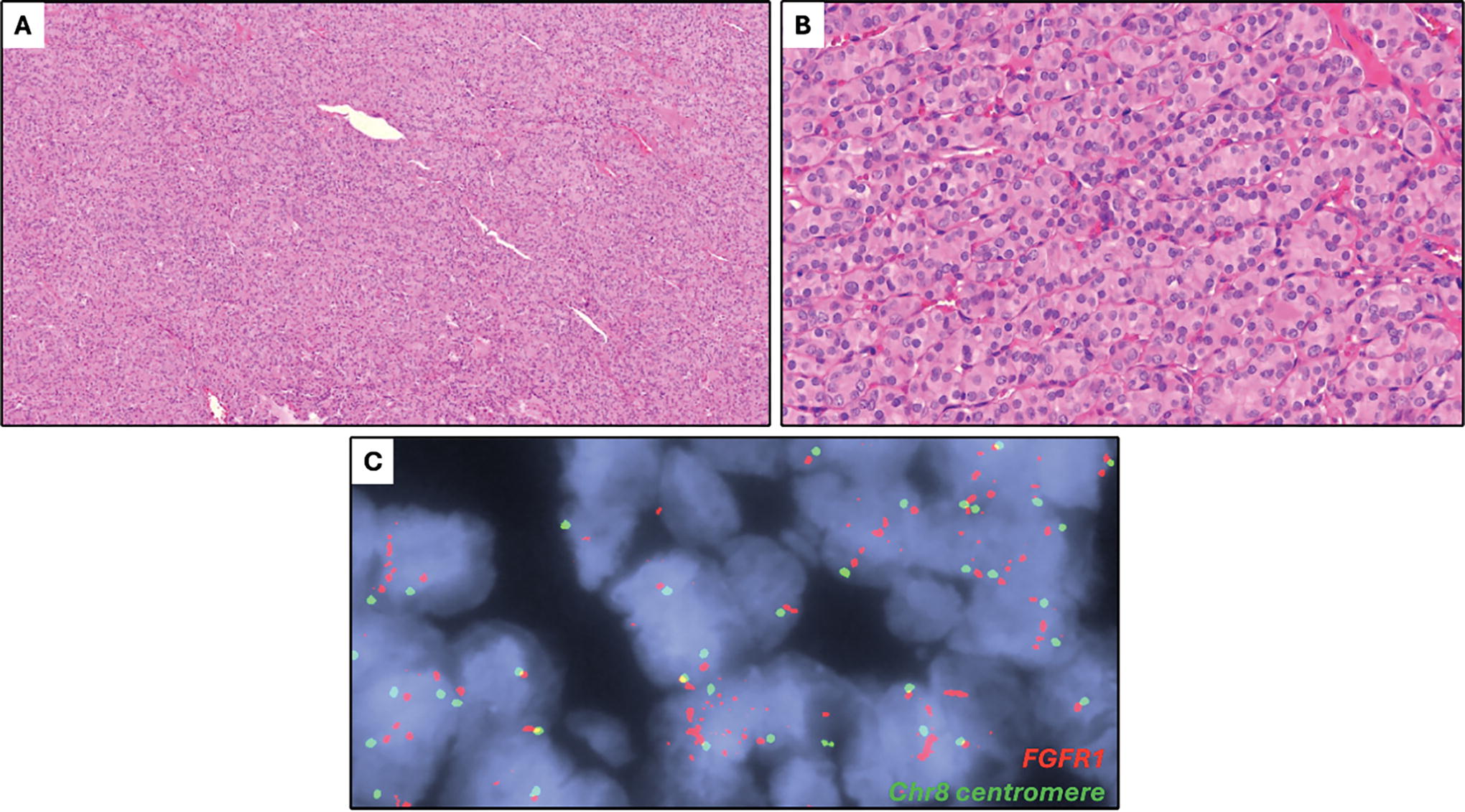
Case 11. A poorly differentiated thyroid carcinoma with a solid growth pattern, as seen on low-power
Clinicopathologic Characteristics of Thyroid Tumors with Primary Driver FGFR Alterations
Likely sampled concurrent follicular thyroid carcinoma with mixed macro- and microfollicular architecture.
AWD, alive with disease; BWH, Brigham and Women’s Hospital; D, at diagnosis; FMI, Foundation Medicine, Inc; MGH, Massachusetts General Hospital; NED, alive with no evidence of disease; NP, not performed; R, at recurrence; RAI, radioactive iodine; U, unknown/unavailable; XRT, external radiation therapy.
FGFR1/2 secondary alterations
Six tumors exhibited FGFR1 or FGFR2 alterations in the background of a known driver of thyroid neoplasia (Table 3). This group was enriched for high-grade tumors (83%) ranging in size from 2.6 to 7.0 cm, including 2 PDTC, a high-grade OTC, an ATC, a thyroblastoma, and an OTC. Most of the alterations seen were sequence variants, including three in FGFR2, and one in FGFR1. Two of the FGFR2 variants, p.Y375C and p.N549K in cases 12 and 16, respectively, showed VAF below 2%; case 12 was an OTC with a near-haploid tumor genome, and case 16 was a PDTC with concurrent TERT promoter C228T variant. The other two secondary FGFR sequence variants identified were FGFR2 p.W290C (12.8% VAF) in a BRAF p.V600E-driven (17.3% VAF) PDTC, and an FGFR1 p.N546K (67.7% VAF) in a high-grade OTC with a near haploid tumor genome. FGFR1 copy number gains were identified in two additional cases, including a DICER1 p.E1705K-driven thyroblastoma (case 14) and a BRAF p.V600E-driven ATC (case 17). NGS also yielded alterations in other genes, including TP53, RB1, PIK3CA, and TERT promoter.
Clinicopathologic Characteristics of Thyroid Tumors with Secondary FGFR Alterations
Also presented in Guilmette J et al. 28
AWD, alive with disease; CRT, chemoradiation therapy; D, at diagnosis; DOD, died of disease; FMI, Foundation Medicine, Inc; MGH, Massachusetts General Hospital; NED, alive with no evidence of disease; NP, not performed; R, at recurrence; RAI, radioactive iodine; U, unknown/unavailable; XRT, external radiation therapy.
Discussion
We present the first cohort of thyroid tumors unified by FGFR alterations, totaling 17, including 11 tumors with presumed primary molecular drivers FGFR1/2 gene fusions and FGFR1 amplifications. FGFR2 fusions consistently involved the canonical exon 17 breakpoint, 29 whereas FGFR1 breakpoints were variable. The high-level amplification of FGFR1 in case 10 that was accompanied by gains in ZNF703 and KAT6A suggests a segmental amplification of chromosome 8p11.2. These alterations are highly likely to be primary drivers as they fit well in the current paradigm of thyroid neoplasia (Fig. 7), with FGFR signaling primarily through the RAS/MAPK and PI3K/AKT pathways. 30,31 Furthermore, they are kinase fusions and CNV that are recurrently seen in PTC and OTC, respectively, the tumors have no other canonical primary drivers of thyroid neoplasia, and the findings build on rare reports of individual thyroid tumors with FGFR alterations, including FGFR2::OFD1, FGFR2::VCL, and TG::FGFR1. 4,5,32 –35
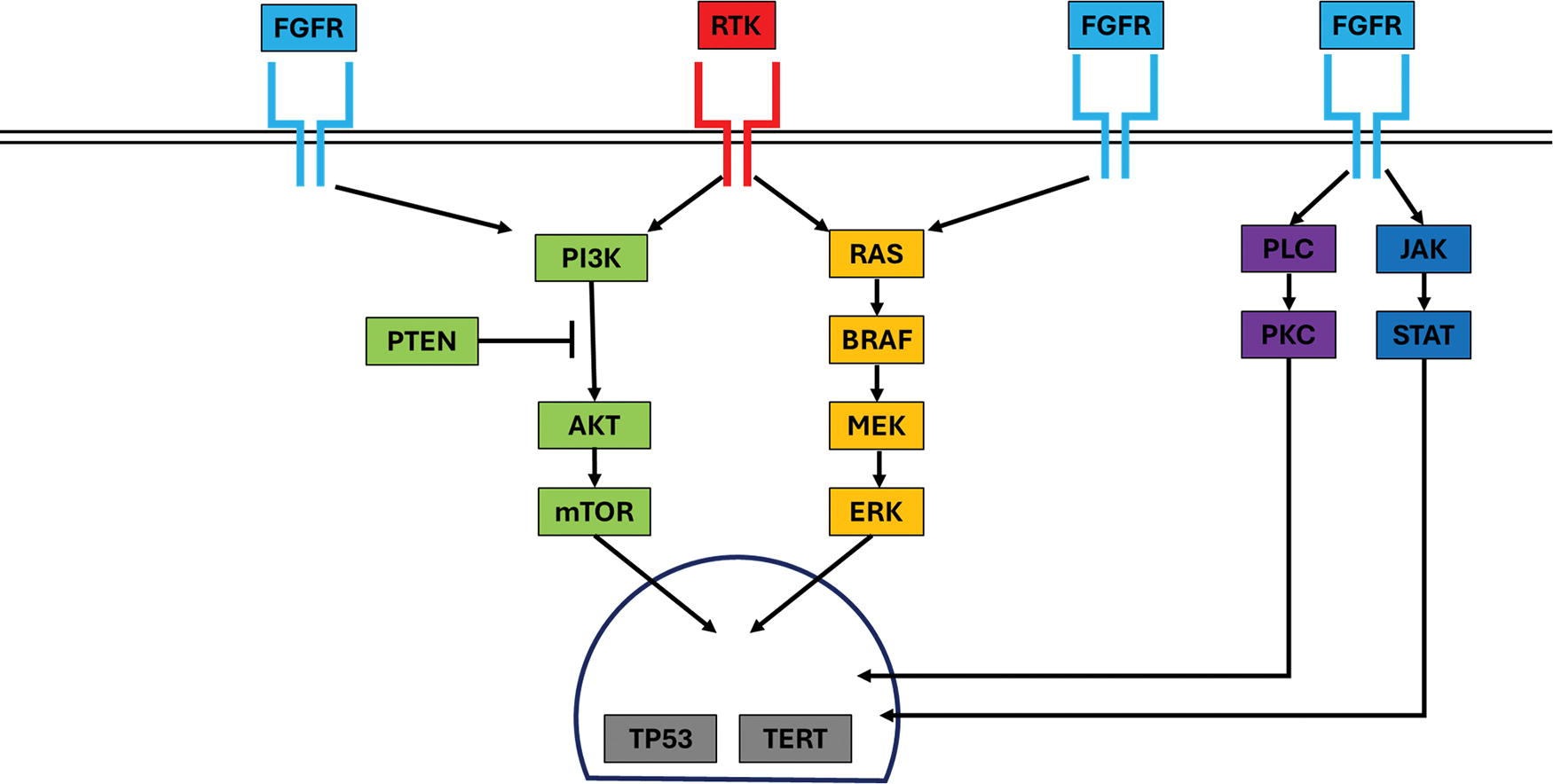
The landscape of molecular findings in thyroid neoplasia, now incorporating the FGFR family of proteins.
The six remaining tumors harbored BRAF p.V600E typical of PTC (cases 15 and 17), haploid genomes in OTC (cases 12 and 13), DICER1 p.E1705K hotspot variant in thyroblastoma (case 14, previously presented in Guilmette et al), 28 and no clear driver was seen in case 12. Cases 12 and 16 had FGFR2 p.Y375C (VAF 1.3%) and p.N549K (VAF 1.1%) alterations at VAF well below those of other variants detected in the respective tumors (5.3–15.8% and 47%), and there was a FGFR1 p.N546K (67.7% VAF) variant seen in case 13, all of which were present in the tyrosine kinase domains (TKD). The variants’ VAF further supports their statuses as secondary alterations, possibly representing subclonal late alterations similar to those seen in TP53 (p.Q331*, p.R280T, p.R273H), PIK3CA p.E545K hotspot, and the C228T TERT promoter variant in this group, or possibly representing enrichment secondary to CNV in OTC for cases 12 and 13.
Another interpretation is that these variants represent lurking or full-fledged facilitators of cancer cell aggression. A study by Raman et al demonstrated FGFR1-mediated adaptive resistance to RET-targeted therapy of a CCDC6::RET-positive thyroid carcinoma, and that dual-inhibition of FGFR and RET signaling improved response to treatment. 21 Also, BRAF p.V600E-driven cell lines were shown to develop resistance to BRAF/MEK inhibition via upregulation of FGFR signaling 36 ; notably, the secondary FGFR1 copy number gains in our study were only observed in the 2 patients with previous targeted therapeutics lenvatinib and dabrafenib. An FGFR3 p.G382R TKD variant appearing secondary in nature has also been described in a co-mutational analysis of 12 BRAF-driven tumors. 37 Posttherapeutic FGFR-mediated resistance in thyroid neoplasia is unsurprising given similar findings following anti-estrogen therapy in breast carcinoma, 38,39 EGFR-specific tyrosine kinase inhibitors in nonsmall cell lung cancer, 40 and paclitaxel in head and neck squamous cell carcinoma. 41 Additional studies examining the dynamic role of these secondary FGFR alterations and their canonical drivers with longitudinal follow-up would yield impactful insights into treating thyroid malignancies.
The study cohort was composed of a breadth of malignancies, including PTC, OTC, PDTC, DHGTC, ATC, and thyroblastoma. Histological review of available slides for FGFR-driven tumors revealed, at this time, that there are no obviously discernible morphological features, in contrast to previous observations in kinase fusion-related thyroid carcinoma. 33,34 For instance, case 1 (Fig. 3) was well-circumscribed, showed extensive papillary architecture, and had a mixture of cells with “papillary-like” nuclear features as well as cells with round, clear nuclei and medium-to-large cytoplasmic compartments. In contrast, case 3 (Fig. 4) showed extensively infiltrative tumor, diffusely “papillary-like” nuclei forming follicles, and only one minute focus of definitively papillary growth. Despite their histological differences, cases 1 and 3 had identical FGFR2::VCL fusions, suggesting a spectrum of morphology and malignant potential in FGFR-driven thyroid neoplasia, and acknowledging the paucity of cases for conclusive comparison.
The FGFR1-4 receptor tyrosine kinases (RTK) are recurrently altered in multiple tumor types. In a large-scale study of 4853 tumors, 343 tumors harbored 360 alterations in FGFR, the most common tumors being urothelial carcinoma followed by breast, endometrial, and ovarian carcinoma, among others. FGFR1 was the most frequently altered, and the observed frequencies of FGFR alterations were 66% amplifications (particularly FGFR1 and FGFR4), 26% sequence variants, and 8% translocations (mostly FGFR2 and FGFR3), 16 concordant with our findings. We observed fusions where FGFR is a 3’ partner (“type I,” e.g., TG::FGFR1), the more typical scenario for RTK fusions, as well as those where FGFR is the 5’ partner (“type II,” e.g., FGFR2 translocations), preserving the TKD and eliminating a C-terminal regulatory domain as seen recurrently in intrahepatic cholangiocarcinoma (FGFR2), glial tumors (FGFR3), and a subset of head and neck squamous cell carcinomas (FGFR3). 18,19,29,42 –46 Discoveries of FGFR driving neoplasia in different tumors is ongoing, including the recently published detection of FGFR1 fusions and amplification in rhabdomyosarcoma. 47 Multiple small molecule inhibitors targeting FGFR genetic alterations are available, including futibatinib, infigratinib, and pemigatinib for cholangiocarcinoma, and erdafitinib for urothelial carcinoma. 17 –20,30,42,43,48,49
A limitation of this study was an enrichment for patients with severe disease, a caveat of using high-confidence data from clinically validated assays in practice where molecular testing is guided by appropriate indications and stewardship. Given the novel association of FGFR alteration with thyroid tumors presented here, further studies exploring the precise mechanisms by which FGFR alterations lead to oncogenesis are warranted. Multiple testing sites created heterogeneity in NGS indications and platforms, as well as in slide and patient record availability. Fortunately, extensive follow-up data were available in many cases, and clinicopathologic data at the time of sequencing were available in all cases. Despite the relatively low prevalence of FGFR alterations in thyroid tumors thus far, the potential for a new class of treatment options via FGFR-targeted inhibitors, which are already in use in other tumors, is an exciting prospect for patients requiring systemic therapy against FGFR-driven thyroid carcinoma, further emphasizing the importance of FGFR variant detection.
Conclusions
To our knowledge, this is the first study presenting a collection of FGFR-altered thyroid tumors, suggesting the relatively sparse knowledge regarding FGFR in thyroid tumors may be due to bias created by a lack of rigorous FGFR analysis during routine testing. Identifying these rare cases will further elucidate clinical parameters from a descriptive format and will, more importantly, identify a cohort of advanced thyroid cancers that may benefit from targeted therapy.
Footnotes
Authors’ Contributions
M.F.S., P.M.S.: Conceptualization, Methodology, Data Curation, Investigation, Visualization, Writing—Original Draft, and Writing-Reviewing and Editing with A.S.F., T.J., D.D.-S., S.S., V.N., L.J.W., G.W.R., J.K.L., B.D., V.N., B.A.A., W.C.F., J.A.B., L.P.L., and A.J.I.: Investigation, Case Contribution, and Writing—Reviewing and Editing. A.S.F.: Conceptualization, Methodology, Data Curation, Investigation, Visualization, Writing—Original Draft, Supervision, and Writing—Reviewing and Editing.
Disclaimer
The content is solely the responsibility of the authors. There are no disclaimers.
Author Disclosure Statement
Tyler Janovitz and Brennan Decker are employed at Foundation Medicine, Inc at the time of publication. Jochen K. Lennerz became employed at Boston Gene Corporation during the time of article production, but no molecular data in the article is acquired from Boston Gene Corporation.
Funding Information
The Max Goodman Fund of the Massachusetts General Hospital Head and Neck Pathology Service provided funding to support publication costs.