
Abstract
Objective:
To evaluate the combined administration of propylthiouracil (PTU) and levothyroxine (LT4) in managing monocarboxylate transporter 8 (MCT8) deficiency and identify optimal therapeutic dosages.
Methods:
This multicenter case series involved 12 male patients with MCT8 deficiency whose parents/guardians consented to PTU and LT4 treatment. Data were collected from January 2008 to June 24, 2024. The study focused on treatment safety and outcomes, analyzing baseline and last encounter biochemical, metabolic, and anthropometric parameters. Statistical analyses included Wilcoxon signed ranks tests and generalized estimated equations to assess effects on thyroid and metabolic markers, and receiver operating characteristics curves to predict optimal dose.
Results:
Patients showed a significant reduction in serum total triiodothyronine (TT3) concentration and TT3/TT4 ratio, with increased serum TT4 and free T4 (fT4) concentrations. The use of PTU effectively reduced TT3 concentration by 25% at an average dose of 6.8 mg/kg/day, while LT4 increased fT4 concentration by 40% from baseline at an average dose of 4.3 µg/kg/day. Thyrotropin concentration was undetectable on treatment. No statistical differences were observed in metabolic and physical parameters between baseline and last encounter overall for the group, but six of eight patients for whom these data were available had an increase in weight (z-score). There were no adverse effects on liver function or granulocyte numbers noted throughout the period of observation.
Conclusion:
Combined treatment with PTU and LT4 normalized serum T3, fT4, and TT4 in patients with MCT8 deficiency. Individualized dose adjustments were crucial for achieving therapeutic goals, indicating the need for personalized treatment plans.
Introduction
Allan–Herndon–Dudley syndrome was described as an X-linked disorder characterized by severe neurodevelopmental impairment, muscular hypotonia, and movement disorders, affecting males from infancy. 1 Sixty years later, the molecular basis of the syndrome was identified as being mutations in the monocarboxylate transporter 8, MCT8 (SLC16A2) gene encoding a specific thyroid hormone (TH) transmembrane transporter. 2,3 As MCT8 is one of the main transporters of thyroxine (T4) into the human brain, local TH insufficiency is responsible for the profound neuropsychomotor impairment in children with MCT8 deficiency, along with characteristic thyroid function tests of low serum T4 and reverse triiodothyronine (rT3) with elevated T3 and normal to slightly elevated serum thyrotropin (TSH) in affected males. These patients have TH deficiency in brain because of impaired transport of TH into the developing brain. Simultaneously, they have excess TH in peripheral tissue, mainly due to increased generation of T3 by deiodination of T4. 4,5 The high serum T3 further upregulates the peripheral tissue deiodinase 1 (D1) with increase in serum T3 5 –7 and decrease in T4 and rT3. There is currently no approved treatment for the neurological or metabolic sequelae of MCT8 deficiency. Conceptually, the ideal treatment would be one where appropriate amounts of TH could be delivered to the brain for normal neural development and at the same time reduce the excessive amount of T3 in peripheral tissues. Provision of an adequate amount of TH to the brain could be accomplished by the use of TH analogues, which are transported into the brain via transporters other than MCT8 or by gene therapy to provide expression of a normal human MCT8 allowing normal TH transport. 8 While human trials are ongoing with the thyroid hormone analogue triiodothyroacetic acid (TRIAC) 9,10 and individual studies have been reported for compassionate use with diiodothyropropionic acid (DITPA), 11 neither treatment is curative and none is able to reverse the neurological deficits when administered postnatally. There are two patient case reports of the use of a combination of levothyroxine (LT4) and propylthiouracil (PTU), which were successful in increasing serum T4 and reducing the T3 on peripheral tissues but with no observed effect on neurological improvement. 12,13 The rationale for this therapy is to provide the highest possible levels of T4 to the brain while preventing peripheral TH excess by inhibiting the conversion of T4 to the active hormone T3 in peripheral tissues. This can be accomplished with PTU, which inhibits D1, which generates T3 from T4 in peripheral tissue without affecting D2, which generates T3 in brain. A reduction of serum T3 would decrease the need for high-calorie intake, requiring frequent feedings and adding a heavy burden on the caregivers. 14
The scarcity of patients due to the rarity of the condition, 1 in 70,000 males 10,12,15 presents significant challenges in conducting large-scale, comprehensive clinical trials, limiting the generalizability of findings and necessitating a continuous search for innovative treatment methods. This multicenter case series, focusing on PTU and LT4 usage in MCT8 deficiency, provides analyses of 12 patients who were unable to obtain DITPA on compassionate basis or TRIAC from a clinical trial or chose not to enroll in that study or their parents/guardians chose to no longer be treated with TRIAC. Both PTU and LT4 are United States Food and Drug Administration-approved drugs for the treatment of hyper and hypothyroidism, respectively, and PTU specifically inhibits D1. 16 The objective of the study was to provide insights into effective management approaches, addressing the significant knowledge gap in the literature regarding combined PTU and LT4 administration for this disorder.
Methods
Study design
This study assesses the use of LT4 and PTU in the treatment of MCT8 deficiency, a rare orphan disease. The study covers the period from January 1, 2008, to June 24, 2024, utilizing data sourced primarily from referring providers associated with the University of Chicago and the University of Miami Health System. Guardians of all individuals provided informed consent for the treatment. Institutional review board (IRB) approval was obtained for the retrospective use of medical data for research purposes in accordance with the University of Miami’s regulations.
Study population
The study was restricted to male patients, reflecting the occurrence of this condition as an X-chromosome-linked disease. Eligible participants had no age limit, with an expectation that the majority of the patients studied would involve children due to the typically shorter lifespan associated with the disease 10 and the current lack of approved treatments. Six of the 12 patients had G-tubes at the time of PTU and LT4 treatment (see Table 1). The earliest treatment was started at 6.3 months.
Characteristics of Subjects with MCT8 Deficiency
Mutation numbering is according to the long form of the MCT8. To convert to the short form subtract 74 amino acids. Initial T4 is reported as % of the lower limit of normal; initial T3 is reported as % of above the upper limit of normal; and TSH is reported as actual value. Subjects ID 4,6,8–12 have not been previously published. Individuals ID 1–3, 17 were previously identified.
d, day; mo, month; TSH, thyrotropin; TT3, total triiodothyronine.
The primary inclusion criterion was to be a patient with a clinically relevant mutation in the MCT8 gene and requested assistance from their health care providers (pediatric endocrinologists and neurologists) for expert assistance in the clinical management of patients with MCT8 deficiency. For obvious reasons, healthy individuals without a pathogenic mutation in the MCT8 gene were not included in this study.
For data collection, we reviewed medical records provided by the patient’s treating physicians. This included records of 12 patients referred from pediatric endocrinologists, geneticists, pediatricians, and pediatric neurologists. Patients were included because their parents/guardians gave permission to participate in the review of the data, agreed to take the medication, and had serum thyroid tests that were consistent with MCT8 deficiency.
The sources of the data were medical records of patients whose parents/guardians had reached out to the study team physicians during their active participation in the care of children with MCT8 deficiency.
This study was conducted following ethical guidelines and received approval from the IRB at the University of Miami 20230297, ensuring compliance with all necessary ethical standards for research involving humans.
Statistical analyses
Continuous variables with normal distribution were expressed as mean ± standard deviation. Normality was assessed using the Shapiro–Wilk test. Continuous variables with asymmetrical distributions were presented as median and interquartile range (IQR, 25th–75th), while categorical variables were presented as absolute and relative frequencies. For comparisons between baseline and last encounter time points, Wilcoxon signed ranks tests were applied.
In this study, treatment adjustments aimed to achieve the highest serum T4 levels (near the upper limit of normal) while reducing serum T3. This was variable among individuals, and treatment with PTU was adjusted to limit further increase of serum T3 as serum T4 levels rose. The increase in PTU dose was stopped when there was no further decrease in serum T3. Various percentage reductions in T3 and increases in T4 were evaluated, and those providing optimal results were set as endpoints. Finally, expected primary endpoints comprised: total T3 (TT3) concentration decreased by 25% from baseline, fT4 concentration increased by 40% from baseline, and TSH was undetectable post-treatment. To analyze individual changes, metabolic parameters were assessed based on their differences from baseline values. Receiver operating characteristic analyses were performed in order to establish the best cutoff point for each dose, either PTU or LT4. Generalized estimated equations (GEE) were performed to analyze the effect of treatment over blood and metabolic markers. We specified separate models for each variable. The GEE models take into account the within-individual correlation and allow us to account for the repeated measurements over time. p Values of <0.05 (two-tailed) were significant, and all data were analyzed on SPSS version 28.0 (SPSS Inc., Chicago, IL, USA) or R statistics.
Results
Twelve males, hemizygous for pathogenic MCT8 mutations detected by Sanger sequencing of a candidate gene or by whole exome sequencing and confirmed by Sanger sequencing, were included, with a follow-up duration ranging from 1 to 136 months (Table 1). At the beginning of treatment, the median age was 18 months (IQR: 9–28 months), and a median of 40 months (IQR: 25–88 months) at the last encounter. The maximum total daily dosage of PTU ranged between 75 and 300 mg, whereas for LT4 it varied between 50 and 250 µg. The number of individual encounters ranged from 4 to 50.
A reduction of TT3 concentration by 10% from the baseline was observed in 12 of the 12 patients within a median time of 50 days; a 25% reduction in TT3 was seen in 7 of the 12 patients, within a median time of 92 days; fT4 concentration increased by 40% in 10 of the 12 patients, within a median time of 27 days; and TSH concentration decreased to undetectable levels in all 12 patients within a median of 27 days.
Comparative delta analyses, assessing the periods before treatment and at the last encounter, revealed a significant reduction in TT3 concentration (p < 0.001) and the TT3/TT4 ratio (p < 0.001), along with a decrease in TSH concentration (p = 0.001). Conversely, there were increases in TT4 (p = 0.005) and fT4 concentrations (p = 0.003) (Table 2, Fig. 1a–f). Hepatic enzymes, specifically alanine aminotransferase, displayed no significant change before (55 ± 37 IU/L) and after treatment (median 42 ± 27 IU/L, p = 0.110, as shown in Fig. 1f), while aspartate aminotransferase levels decreased from before (55.9 ± 22 U/L) to after treatment (40.2 ± 16 U/L, p = 0.013, as shown in Fig. 1g).
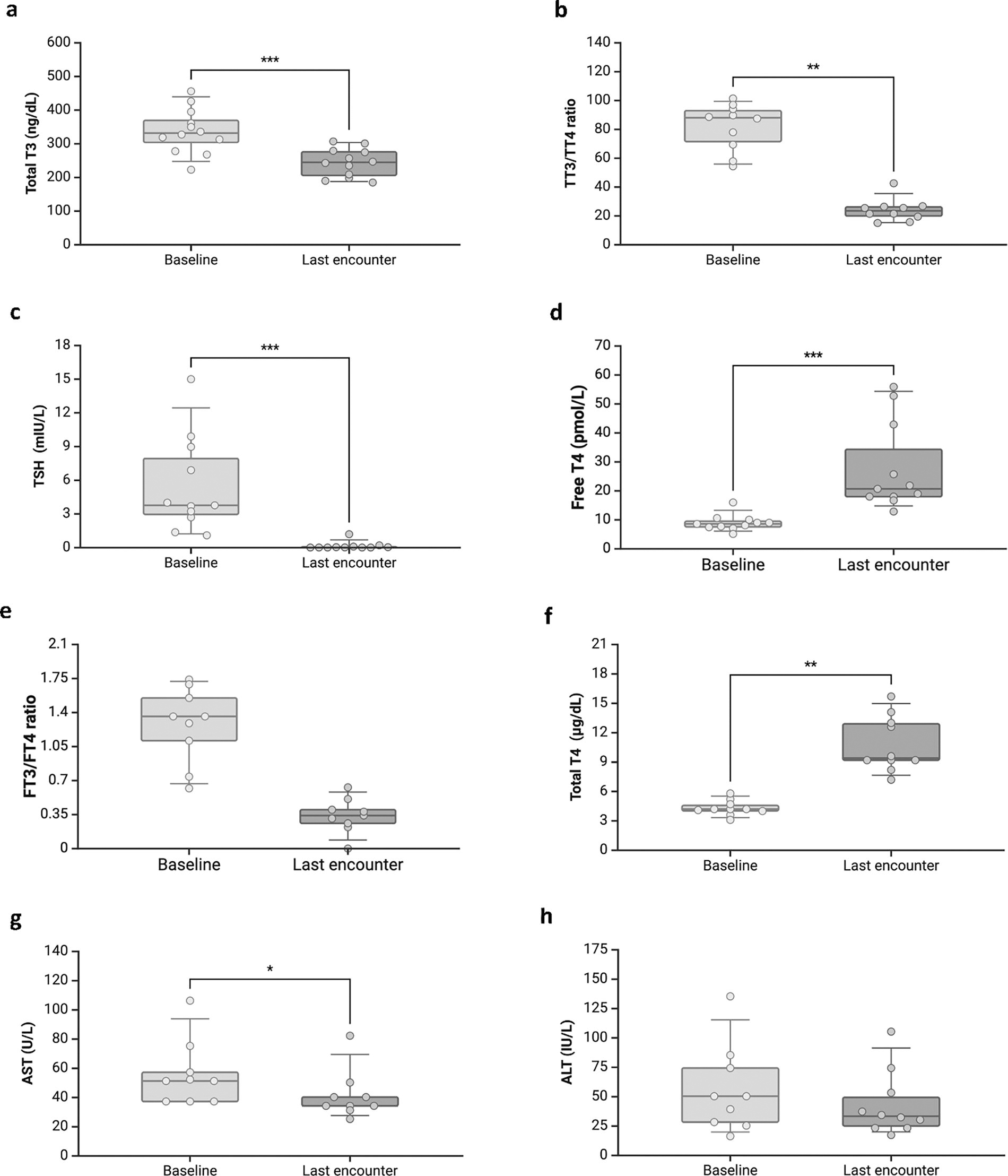
Boxplot showing the comparison between baseline and last encounter after treatment.
Comparative Delta Analysis of Metabolic and Biochemical Parameters Between Baseline and Last Encounter in Patients with MCT8 Deficiency Treated with PTU and LT4
CK, creatine kinase; HR, heart rate; LT4, levothyroxine; MCT8, monocarboxylate transporter 8; PTU, propylthiouracil; SDS, standard deviation score; SHBG, sex hormone binding globulin.
Changes in metabolic and physical parameters
Upon evaluation of metabolic parameters, notable interindividual variability was observed (Fig. 2). The difference in weight was analyzed separately for patients with feeding tubes and those without. Among patients with complete weight information available and without feeding tubes, all four showed an increase in weight, measured standard deviation score (SDS) (from a baseline of −2.18 ± 1.2 to −1.7 ± 1.06 at the last encounter, p = 0.068). In the group with feeding tubes, two out of four patients exhibited an increase in SDS weight, while the remaining two patients showed a decrease. Comparisons of mean values between baseline and last encounter measurements revealed no statistically significant differences (Table 2).
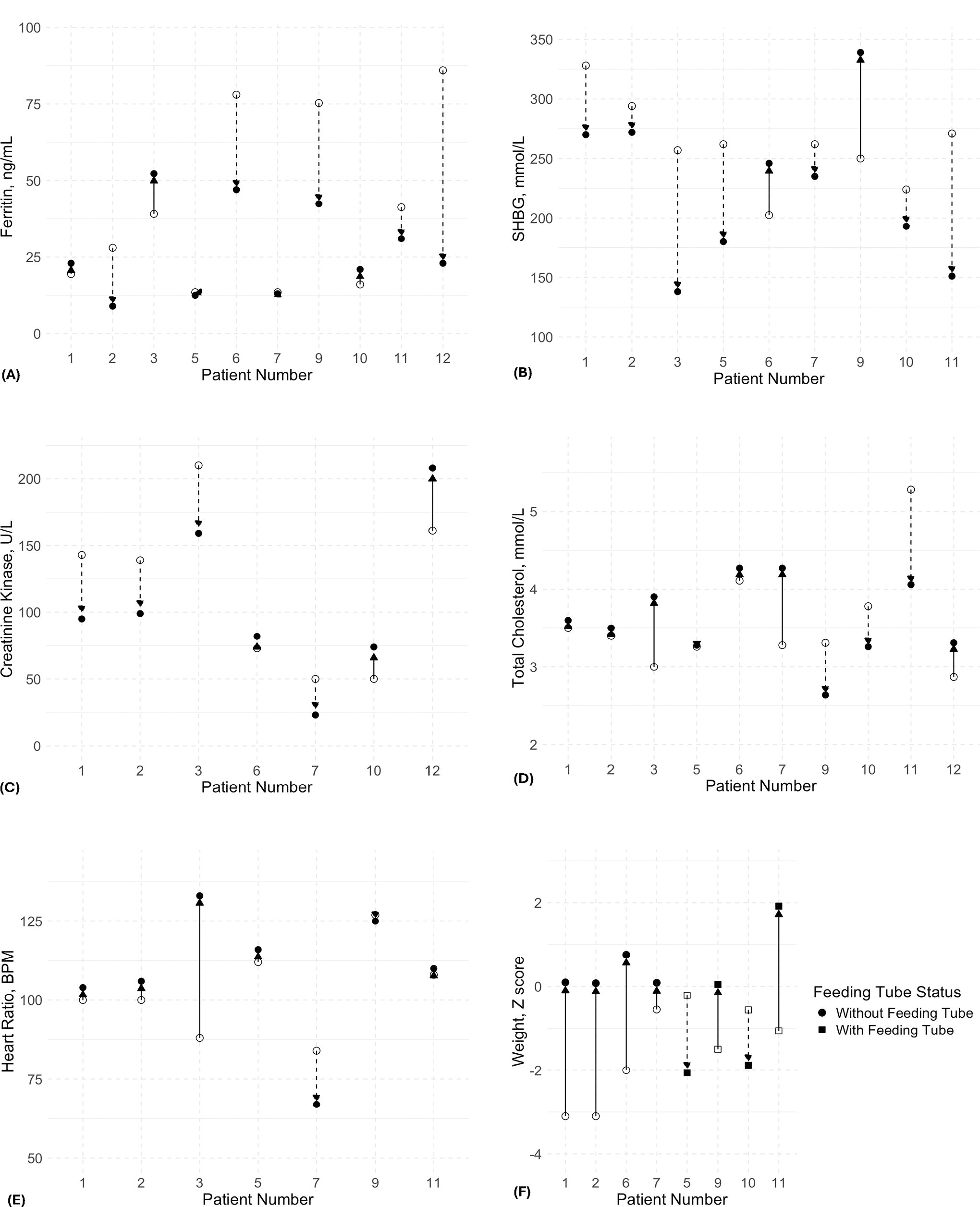
Representation of the progression of clinical and biochemical parameters in patients over the course of treatment. Of note, these parameters were not available in all 12 patients in this study. Each patient’s baseline measurements are depicted by the open symbol, while the change up to the last encounter is represented in the solid symbol. Arrows indicate the direction of change from baseline to the last encounter, with upward arrows signifying an increase and downward arrows a decrease.
Predicting optimal therapeutic dose
The dosages of PTU and LT4 were individually tailored to achieve specific endpoints: reduction in TT3 concentration, increase in fT4 concentration, and undetectable TSH levels. PTU effectively reduced TT3 concentration by 25% from baseline at an average total daily dose of 6.8 mg/kg, achieving an area under the curve (AUC) of 0.670 (CI: 0.560–0.780; p = 0.007) (Supplementary Fig. S1). This occurred despite the LT4-mediated increase in T3. In contrast, LT4 successfully increased fT4 concentration by 40% from baseline at an average daily dose of 4.3 µg/kg achieving an AUC of 0.947 (CI: 0.878–0.999; p < 0.0001) (Supplementary Fig. S2). Furthermore, TSH levels became undetectable with a total daily LT4 dose of 4.4 µg/kg (AUC: 0.898; CI: 0.811–0.986; p < 0.0001) (Fig. 3).
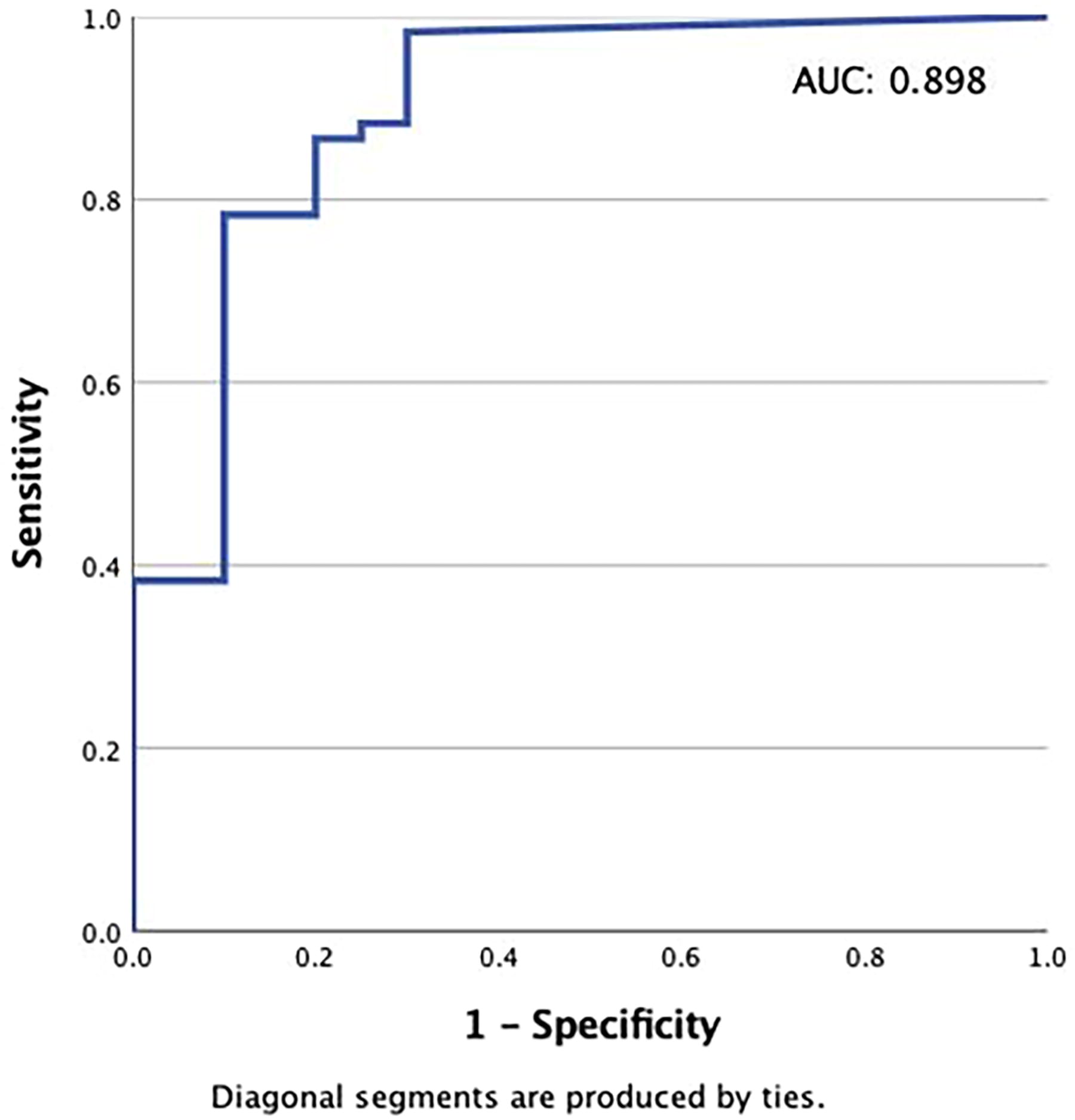
Levothyroxine (LT4) dosing for targeted TSH suppression. The dose–response relationship of LT4 and undetectable TSH levels. Receiver operating characteristic (ROC) curve that determines the optimal total daily dose per kg of LT4 that correlates with achieving undetectable TSH levels.
Discussion
In this multicenter case series study, we investigated the effects of PTU and LT4 treatment in patients with MCT8 deficiency. This genetic disorder, linked to mutations in the MCT8 gene on the X chromosome, leads to profound psychomotor delays in affected males generally with inability to walk, talk, or care for themselves, while heterozygous females are asymptomatic. 10 Our findings indicate that treatment with PTU and LT4 effectively reduced serum total T3 concentration compared to the untreated baseline, despite the fT4 mediated T3 increase. The serum total T4 and fT4 concentrations increased with the LT4 treatment. Diagnosis in male infants is often delayed beyond the neonatal period because neonatal screening is based on the measurement of TSH. Thus, unless there is a known family history, the asymptomatic carrier mothers are not suspected of possibly delivering an affected male. Further, affected male newborns commonly develop without concern until approximately six months of age, when the delay in developmental milestones and significant hypotonia become apparent to parent and physician, and either the pathognomonic thyroid tests (low T4 and rT3, high T3, and normal to high TSH) suggest the diagnosis or direct genetic analysis reveals a mutation in the MCT8 (SLC16A2 gene). 6,18 While newborn screening programs measuring all serum iodothyronines could diagnose these children at birth 19 and start treatment early, such routine screening is not yet available.
Patients with MCT8 deficiency present a unique therapeutic challenge due to the combination of brain TH deficit and peripheral tissue TH excess. Current therapeutic options are limited, with TH analogues such as TRIAC and DITPA being used to modulate thyroid status. However, these treatments are not universally available, both effectively reduce the serum T3 level. Our study investigates the therapy using PTU and LT4, which are more accessible and cost-effective than the other two options. The hypotonia and neurodevelopmental delays have no currently known treatment except for intensive physical and occupational therapy to maximize the plasticity and development of the nervous system. The peripheral tissue TH excess on the contrary is more amenable to treatment. The rationale of using TH analogues is to decrease the serum T3 concentration. TRIAC accomplishes this by reduction of TSH resulting in decreased secretion of both T4 and T3. 20 DITPA decreases serum T3 by inhibiting D1. As no data are available to what extent TRIAC and DITPA in the amounts used reach the brain, there is concern regarding the observed further reduction of serum T4 levels. The latter is present in brain of MCT8 deficient albeit in reduced amounts. 21 Thus, it is expected that an increase in serum T4 will provide more T4 to the brain even in the setting of MCT8 deficiency, as demonstrated in an MCT8 deficient treated in utero with high doses of LT4. 22 This is particularly important as T4 is more available than T3 to the brain being better transported across the blood–brain barrier 23 thus suggesting further brain TH deficit with TRIAC treatment. A retrospective cohort study on MCT8-deficient patients demonstrated that long-term TRIAC treatment reduced serum T3 concentrations, improved body weight, decreased heart rate, and lowered sex hormone binding globulin concentrations, with no significant change in creatine kinase levels and no severe drug-related adverse events. 24
While there has been no head-to-head trial comparing the efficacy of these three treatments, the purpose of this report is to provide additional information on an alternative therapy to DITPA and TRIAC, which are currently only available via a trial, whereas PTU and LT4 are readily available at low cost throughout the world. Furthermore, one concern expressed by collaborating physicians in the care of these patients was regarding the hepatotoxicity of PTU. While it was recommended that its use in the pediatric population be discontinued 25,26 or used with caution, 27 a recent large meta-analysis in pregnancy showed that it was safer than methimazole. 28 The rationale for using PTU in this study was that there is no other option to reduce the serum T3 while increasing the T4 concentration. The use of methimazole in children with MCT8 deficiency is not an option because it lacks the effect of inhibiting peripheral tissue D1, which is characteristic only of PTU. Providers and the parents of affected children need to weigh the risk-benefit ratio in the use of these medications. In our limited study, we did not find any evidence of liver toxicity; however, close monitoring of liver function tests is indicated given the concern and the relatively high doses of PTU used. Should hypogranulocytosis or increase in liver function tests occur, we recommend stopping the treatment. If only mild, repeating the challenge in a month or continuing cautiously as long as leucocyte and liver abnormalities do not continue to change.
Limitations
Retrospective case series studies often depend on the accuracy and completeness of medical records, which can vary. The number of patients in this study is not sufficiently powered to detect significant differences in peripheral tissue markers of TH action due to the variable baseline. Furthermore, the length of treatment was variable. Direct comparisons to other treatments such as DITPA and TRIAC are not possible in this report. With respect to weight changes, we were unable to control calorie intake and do not know if weight was maintained with less food as the T3 decreased.
Strengths
This design allowed for a comprehensive analysis of treatment approaches and patient outcomes over nearly two decades, providing a unique and extensive perspective on the management of this rare genetic disorder. The inclusion of data from various referrals—pediatric endocrinologists, geneticists, pediatricians, and pediatric neurologists—enriched the study, offering a broad view of the therapeutic landscape and the evolution of care practices for patients with MCT8 deficiency. The retrospective nature of the study facilitated an in-depth examination of long-term treatment efficacy and patient progression while adhering to ethical standards of patient confidentiality and data integrity.
The sources of the data were highly specific and included medical records of patients provided by the treating physicians during their active participation in the care of these children with MCT8 deficiency. This participation extended beyond direct medical care to advising other endocrinologists and acting as consultants for parents/caregivers and peers from the endocrine field. The study physicians have also been involved as advisors to the AHDS/MCT8 patient advocacy group.
Conclusions
Our study presents the complexity of treating MCT8 deficiency and highlights the combined PTU and LT4 administration as promising alternative to current TH analogue therapies to modulate the peripheral tissue TH excess while providing higher concentration of T4. Our analysis can be used as a guideline for physicians caring for these patients regarding the average daily PTU and LT4 doses to effectively reduce TT3 levels while increasing the fT4 levels depending on the age of the patients when starting this treatment. Ongoing research and collaboration among specialists are essential to improving outcomes for patients with this challenging condition.
Data statement
The datasets generated and/or analyzed during the current study are available from the corresponding author upon reasonable request and subject to any necessary ethical approvals.
Footnotes
Acknowledgments
The authors thank the patients and guardians for participation in the research and the following physicians who provided medical care and the follow-up of the treatment: Dr. Charles Verge, Sydney Children’s Hospital, Sydney, Australia; Jill Hamilton, The Hospital for Sick Children, Toronto, Ontario, Canada: Dr. Jeffrey Kaplan, Westchester Medical Center and Nyack Hospital, Monroe, NY; Drs. Omar Ali and Peter Wolfgram, Medical College of Wisconsin, Milwaukee, WI; Dr. Cunita Cheruvu, Goryeb Children’s Hospital, Morristown, NJ; Dr. Margaret Siska, St. Louis University Health Science Center, St. Louis, MO; Dr. Sobenna George Emory University School of Medicine, Atlanta, GA; Drs. Stacy Rustico and Stecey Schmiedecke, Naval Medical Center, San Diego, CA; and Dr. Pamela SW Smith, Phoenix Children’s Hospital, Phoenix, AZ.
Authors’ Contributions
All authors met authorship criteria and participated sufficiently in the work. All authors certify that this material has not been submitted or published before. All authors confirm that the research meets the ethics guidelines. All authors have approved the final article and agree to be accountable for the work. R.E.W. participated in the performance of the analysis, designed the study, discussed the results, participated in the writing of the article, and gave final approval to the article. J.R.N.L. reviewed the results, provided statistical analysis, participated in the writing of the article, and gave final approval of the article. A.M.D. was involved in the study design, discussed the results, participated in the writing of the article, and gave final approval to the article. M.S.I. provided analysis and confirmation of genotypes. K.H. provided regulatory input, discussed the results, participated in the writing of the article, and gave final approval to the article. S.R. was involved in the study design, discussed the results, participated in the writing of the article, and gave final approval to the article.
Author Disclosure Statements
R.E.W. declares there are no conflicts of interest but notes that he is on the advisory board of Prizm Therapeutics. J.R.N.L. declares there are no conflicts of interest and has nothing to declare. M.S.I. declares there are no conflicts of interests and has nothing to declare. K.H. declares there are no conflicts of interest but notes that he is on the advisory board of Prizm Therapeutics. A.M.D. declares there are no conflicts of interest and has nothing to declare. S.R. declares there are no conflicts of interest and has nothing to declare.
Funding Information
This work was supported by grants from the National Institutes of Health, USA, DK
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2