
Abstract
Background:
Two selective RET inhibitors (RETis) are effective in treating
Methods:
We conducted a retrospective cohort study of patients with hMTC treated with a selective RETi at a tertiary cancer center. The primary outcome was overall response rate using Response Evaluation Criteria in Solid Tumors (RECIST) v1.1. Secondary end points included overall survival (OS), progression-free survival (PFS), biochemical response rate, and safety.
Results:
We identified 23 evaluable patients as follows: 15 (65%) multiple endocrine neoplasia (MEN)2A and 8 (35%) MEN2B. Median age at start of RETi was 51 years (range, 15–79). All patients had distant metastases, and 52% (12/23) had received prior systemic therapy (median = 1, range, 0–3). Patients were treated with selpercatinib (n = 13) or pralsetinib (n = 10), 57% (13/23) within a clinical trial. Median duration of RETi was 25 months (range, 3–72) with 11/23 (48%) patients remaining on drug at data cutoff due to an ongoing response. Median duration of follow-up was 49 months (range, 9–72). Best radiographical response was partial response in 18 (78%) and stable disease in 5 (22%) patients. Median OS was 51 months (confidence interval, 40.5–61.3); median PFS was not reached. Most common adverse events (AEs) were increased alanine aminotransferase (ALT) (48%) and aspartate aminotransferase (26%), dry mouth (39%), QT interval prolongation (39%), fatigue (35%), and hypertension (26%). AEs led to dose reductions in eight (35%) patients. No grade 5 treatment-related AEs occurred. While the germline nature of the RET pathogenic variant in hMTC could hypothetically result in increased drug-related toxicity, the incidence of most AEs, other than grade 1–2 ALT elevation and QT interval prolongation, was comparable to published clinical trials.
Conclusions:
In patients with advanced hMTC, selective RETis appear safe and effective with outcomes similar to clinical trial cohorts, which mostly comprised patients with sMTC. Duration of response and AE profile was similar to sMTC, although longer follow-up and larger patient numbers are needed to confirm this.
Introduction
Medullary thyroid carcinoma (MTC) is a rare subtype of thyroid cancer that arises from the parafollicular, calcitonin-secreting C cells. About one quarter of MTCs in adults are hereditary
1,2
and caused by an autosomal dominantly-inherited, activating mutation in the
In addition to a nearly 100% lifetime penetrance of MTC, the MEN2A clinical spectrum also includes pheochromocytoma, primary hyperparathyroidism, cutaneous lichen amyloidosis, and Hirschsprung disease. 3,6 Over 100 RET alterations have been identified in patients with MEN2A, and there are strong genotype–phenotype correlations. 6,7 MEN2B accounts for only 5% of hMTC, but it leads to early-onset disease that can present during infancy. It is also characterized by pheochromocytoma, skeletal abnormalities, intestinal dysmotility leading to megacolon, mucosal neuromas, and ophthalmological abnormalities. 4,6,8 MEN2B is due to the germline RET p.M918T pathogenic variant in almost all patients.
Up to 15% of patients with MTC have distant metastases at initial presentation, 6,9 which is associated with a 10-year survival rate of 40%. 10 Over the past 15 years, notable progress has been made in the treatment of metastatic RET-altered MTC. 11 While the multikinase inhibitors vandetanib and cabozantinib are effective in treating advanced MTC, they are associated with notable toxicities due to off-target activity, specifically vascular endothelial growth factor receptor (VEGFR) inhibition. 12 –14 Recently, two highly selective small molecules with potent anti-RET activity, selpercatinib and pralsetinib, have been approved by regulatory agencies for the treatment of RET-altered tumors. These drugs showed significant efficacy and limited toxicity in patients with advanced RET-altered thyroid cancers in phase I/II clinical trials. 15 –18 Selpercatinib also demonstrated superior progression-free survival (PFS) and tolerability compared with cabozantinib or vandetanib in RET-altered MTC in a phase III randomized trial (LIBRETTO-531). 19 However, none of these studies delineated responses between hereditary and sporadic cases of MTC.
hMTC presents at a younger age and is more often multifocal compared with sporadic MTC. 1,2,20 In addition, most hMTCs harbor a RET mutation within the cysteine-rich domain of the RET receptor (exons 10 and 11), while somatic MTCs are most commonly driven by a mutation in the tyrosine kinase domain (exon 16; p.M918T). 21 Furthermore, certain conditions associated with the MEN2 syndromes, including gastrointestinal dysmotility, eye dryness, or pheochromocytoma-associated hypertension, overlap with the side effect profile of the selective RET inhibitors (RETis). Thus, genotypic and phenotypic differences between hMTC and sporadic MTC (sMTC) raise the question as to whether or not they respond similarly to RETi or if the side effect profile may be worse in those with MEN2 who harbor a RET pathogenic variant in the germline. Therefore, we conducted a retrospective, single-center cohort study aiming to assess the efficacy and safety of RETi in patients with advanced hMTC.
Materials and Methods
Study population
This single-center retrospective cohort study was conducted at the University of Texas MD Anderson Cancer Center and approved by the Institutional Review Board. The need for patient consent was waived by the ethics board under protocol RCR04-0933. All patients with advanced hMTC initiating therapy with a RETi between March 1, 2017 and June 30, 2023 were eligible, including patients treated on a published clinical trial or as standard of care. Patients treated on a neoadjuvant selpercatinib clinical trial (NCT04759911), 22 which is still in progress, were excluded. The data cutoff was January 15, 2024, allowing at least two restaging visits for each patient.
Outcomes and study assessments
The study’s primary outcome was overall response rate (ORR), defined as complete responses (CRs) plus partial responses (PRs), according to the Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1, 23 as assessed by our radiologists. Secondary endpoints included overall survival (OS), PFS, biochemical response rate, and safety. OS was defined as the time between the first RETi dose and death from any cause; PFS was defined as the time between the first RETi dose and disease progression or death. Patients who had a continued response or were still alive at the data cutoff were censored at last staging visit or last follow-up for PFS and OS analyses, respectively. Biochemical responses for serum calcitonin and carcinoembryonic antigen (CEA) were defined as a biochemical CR if the levels normalized, biochemical PR if a decrease of ≥50% from baseline occurred, biochemical stable disease (SD) if a decrease or an increase of <50% from baseline occurred, and biochemical progressive disease (PD) if an increase of ≥50% occurred (each maintained for ≥4 weeks). 13 Biochemical response rate was reported as the proportion of patients with a biochemical CR or PR. Adverse events (AEs) were graded according to the U.S. National Cancer Institute Common Terminology Criteria for Adverse Events (version 5.0). 24 Demographics, molecular analyses, previous therapies, AEs, and survival data were extracted from the patients’ electronic medical records.
Statistical analyses
Demographic and treatment characteristics were presented as medians with ranges for continuous variables and as numbers with percentages for categorical data. The Kaplan–Meier method was used to analyze PFS and OS endpoints. Estimates of duration of follow-up were based on the inverse Kaplan–Meier method. Hazard ratios (HRs) and two-sided confidence intervals (CIs) were calculated through univariate Cox proportional hazard regression. A two-sided p < 0.05 indicated statistical significance. SPSS Statistics for Macintosh, Version 29.0 (Released 2022. Armonk, NY: IBM Corp) was used for statistical analyses.
Results
Patients
During the defined study period, a total of 175 patients with RET-altered MTC treated with a selective RETi were seen at our institution, 24 (14%) of whom had hereditary disease (Supplementary Fig. S1). Twenty-three patients met the inclusion criteria for the present study. Fifteen patients (65%) had MEN2A, and 8 (35%) had MEN2B. Sixty-one percent of patients were female. Baseline characteristics for both groups are detailed in Table 1. The most frequent RET variants in patients with MEN2A were in codons 620 (27%), 634 (27%), and 804 (27%), whereas all patients with MEN2B had the germline RET p.M918T pathogenic variant. At the start of RETi therapy, median age was 51 years (range, 15–79), and all patients had distant metastases. Not surprisingly, MEN2B patients were younger at age of hMTC diagnosis and RETi start. No patients had known brain metastases prior to starting therapy, although only six (26%) had baseline brain imaging. Most patients had an Eastern Cooperative Oncology Group performance status of 0 or 1. About half of the patients (52%) had received at least one previous line of systemic therapy, including cabozantinib in two patients (9%), vandetanib in four patients (17%), and both cabozantinib and vandetanib in three patients (13%). Median time from hMTC diagnosis to RETi therapy was 9.0 years (range, 1.5 months–52 years). Thirteen patients (57%; 6 on trial) were treated with selpercatinib and 10 (43%; 7 on trial) with pralsetinib.
Baseline Characteristics of 23 Patients with Hereditary Medullary Thyroid Carcinoma Treated with a Selective RET Inhibitor
MTC, medullary thyroid carcinoma; yrs, years; no, number; ECOG, Eastern Cooperative Oncology Group; RETi, RET inhibitor; MEN, multiple endocrine neoplasia; NA, not available.
Overall efficacy
Median duration of treatment with a RETi was 24 months (range, 3–72). In the overall cohort, ORR was 78%: 18 patients had a PR (78%), while 5 had SD (22%), and none had a CR. Efficacy appeared similar in patients with MEN2A and MEN2B, as detailed in Figure 1A. ORR was also similar whether patients were treated with selpercatinib (85%) or pralsetinib (70%) (p = 0.618). With a median follow-up of 49 months, median PFS in the overall cohort was not reached, and the 24-month PFS rate was 66% (Table 2). Efficacy was observed regardless of previous systemic therapy exposure as follows: ORR was 82% in treatment-naive patients compared with 75% in those who received ≥1 previous systemic therapy (p = 1.00). There was also no significant difference in PFS between treatment-naive and previously treated patients (p = 0.853; Supplementary Fig. S2A), MEN2A and MEN2B patients (p = 0.252; Supplementary Fig. S2B), or selpercatinib and pralsetinib (p = 0.663; Supplementary Fig. S2C).
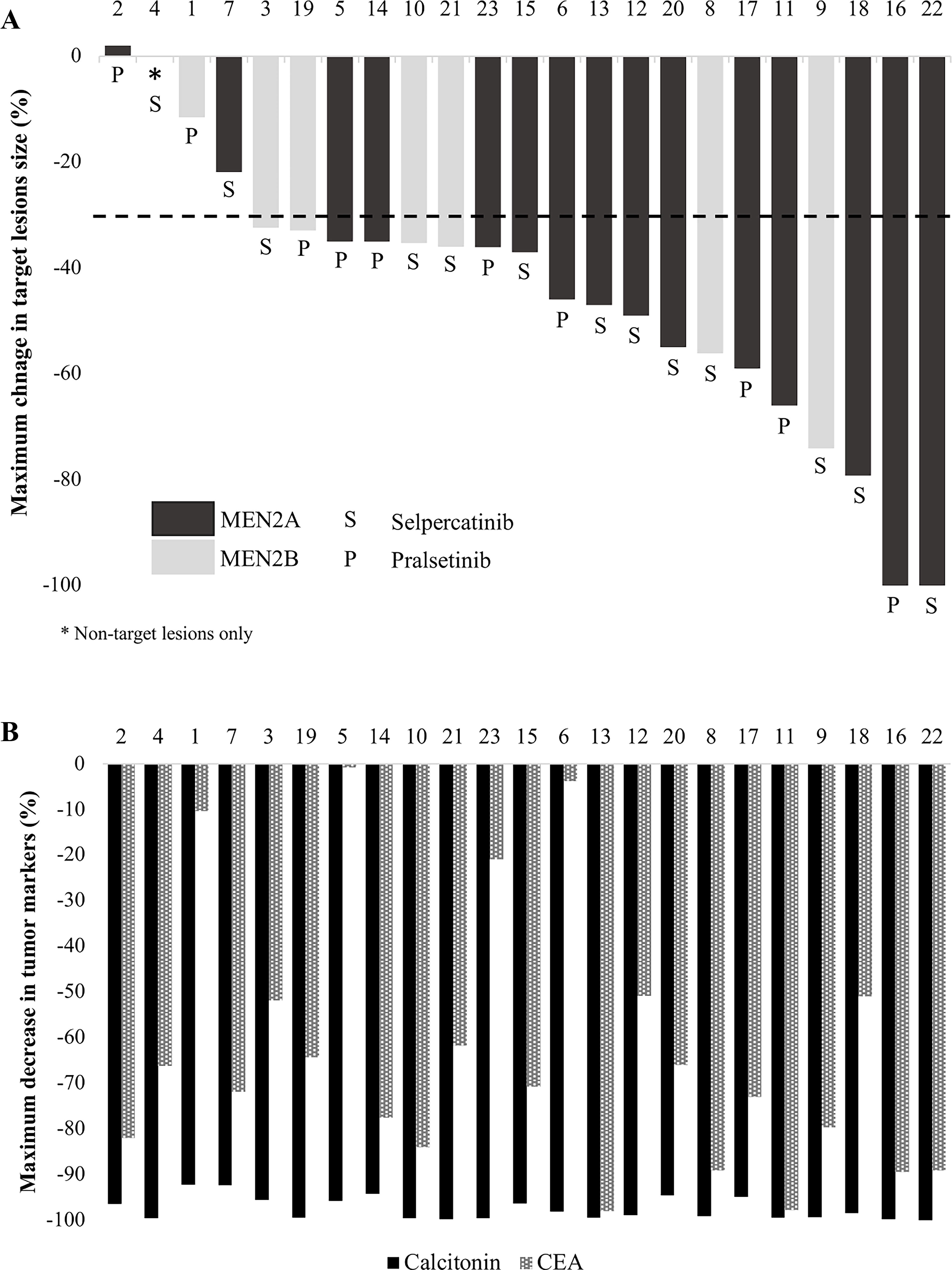
Maximum change in target lesions
Summary of Response to a Selective RET Inhibitor in 23 Patients with Hereditary Medullary Thyroid Carcinoma
No, number; mos, months; CI, confidence interval; NR, not reached.
Median OS from start of the RETi was 51 months (CI, 40.5–61.3). There was no significant difference in OS between MEN2A and MEN2B patients as follows (Fig. 2): median OS was 39 months (CI, 12.8–65.2) in patients with MEN2A and was not reached in patients with MEN2B (p = 0.543). Univariate Cox regression analysis did not identify any factor as a significant predictor for mortality, including sex (HR 1.71; p = 0.468), age at diagnosis (HR 1.01; p = 0.569), prior systemic therapy (HR 1.55; p = 0.553), bone (HR 0.39; p = 0.271) or liver metastases (HR 1.05; p = 0.968) at the start of RETi, choice of RETi (HR 0.96; p = 0.956), or time from diagnosis to start of RETi (HR 1.00; p = 0.890).
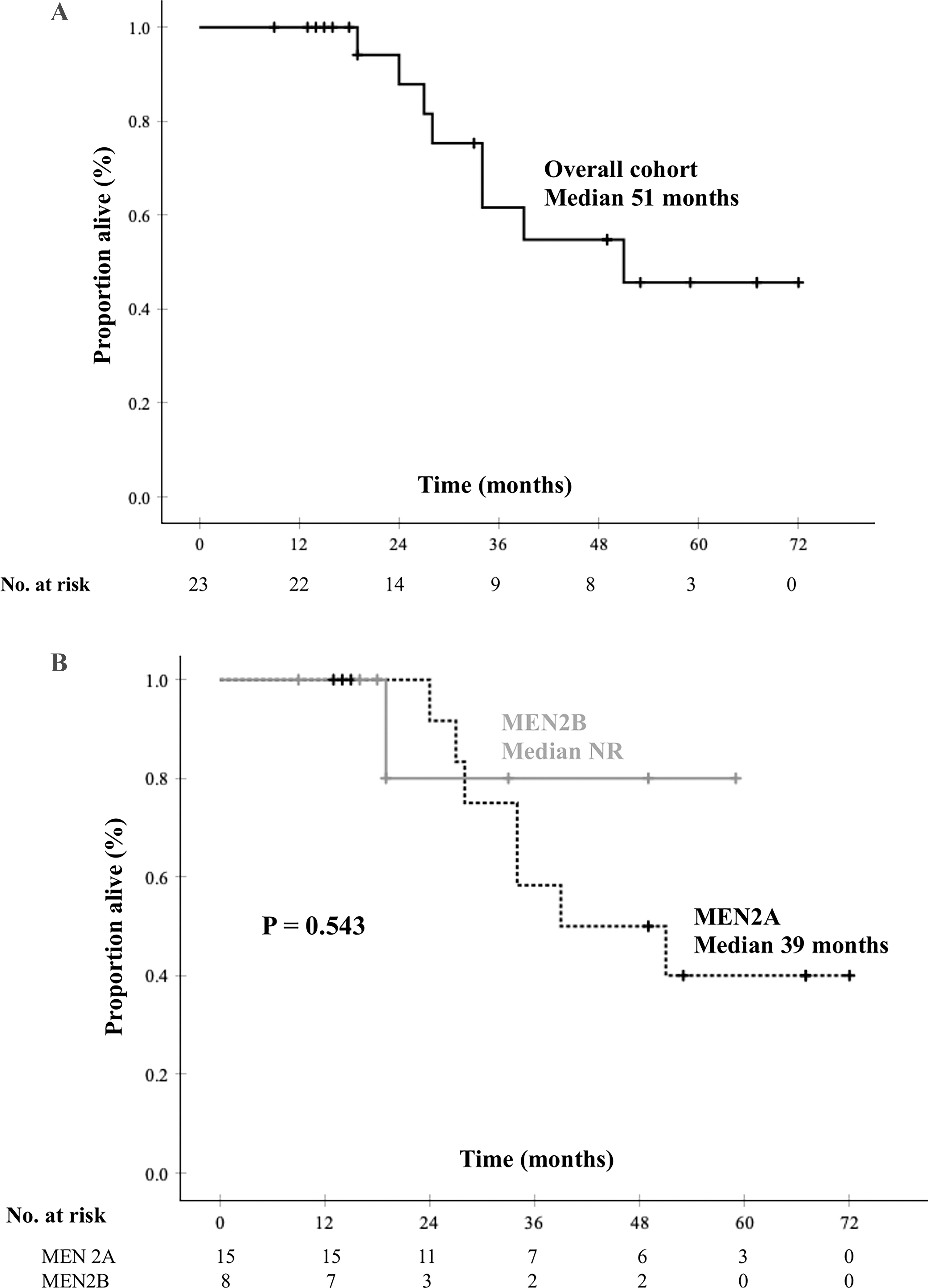
Kaplan–Meier estimates of overall survival (OS) in the entire cohort of hereditary medullary thyroid carcinoma patients treated with a selective RET inhibitor
Biochemical responses
All patients had elevated baseline levels of calcitonin and CEA. Biochemical response rate was 100% for calcitonin and 83% for CEA. Figure 1B summarizes the maximal declines in tumor markers. Median maximal decrease in calcitonin was 98.9% (range, 92.1–99.9), and median maximal decrease in CEA was 70.7% (range, 0.8–98.0); there was no difference between MEN2A and MEN2B patients (p = 0.357 and p = 0.825, respectively). Importantly, the biochemical response did not necessarily correlate with the structural response (Fig. 1).
Patients’ disposition
At data cutoff, 11 patients (48%) remained on a RETi with an ongoing response to therapy (Fig. 3). Eight patients had disease progression at a median time of 20.5 months (range, 13–32 months), and seven of these had repeat molecular testing available at time of progression (Table 3). On-target acquired somatic RET mutations were observed in 43% of patients as follows: RET solvent-front G810 mutations in two patients, including one patient in whom liquid biopsy showed both RET p.G810C and p.G810R mutations, as well as a RET p.M918T variant, and a RET p.V804M gatekeeper mutation in another patient. Bypass resistance alterations were identified in 29% of patients as follows: an EML4::ALK fusion in one patient and an FGFR3 variant in another. TP53 and RB1 alterations were observed in 29% and 14% of the tested tumors, respectively. Eight patients had died at data cutoff, including all seven patients in Table 3. Of the six patients for whom the cause of death was known, five died due to disease progression and one died of respiratory failure as a complication from cryoablation of a rib metastasis. Table 4 further details the clinical course of the eight patients who have died.
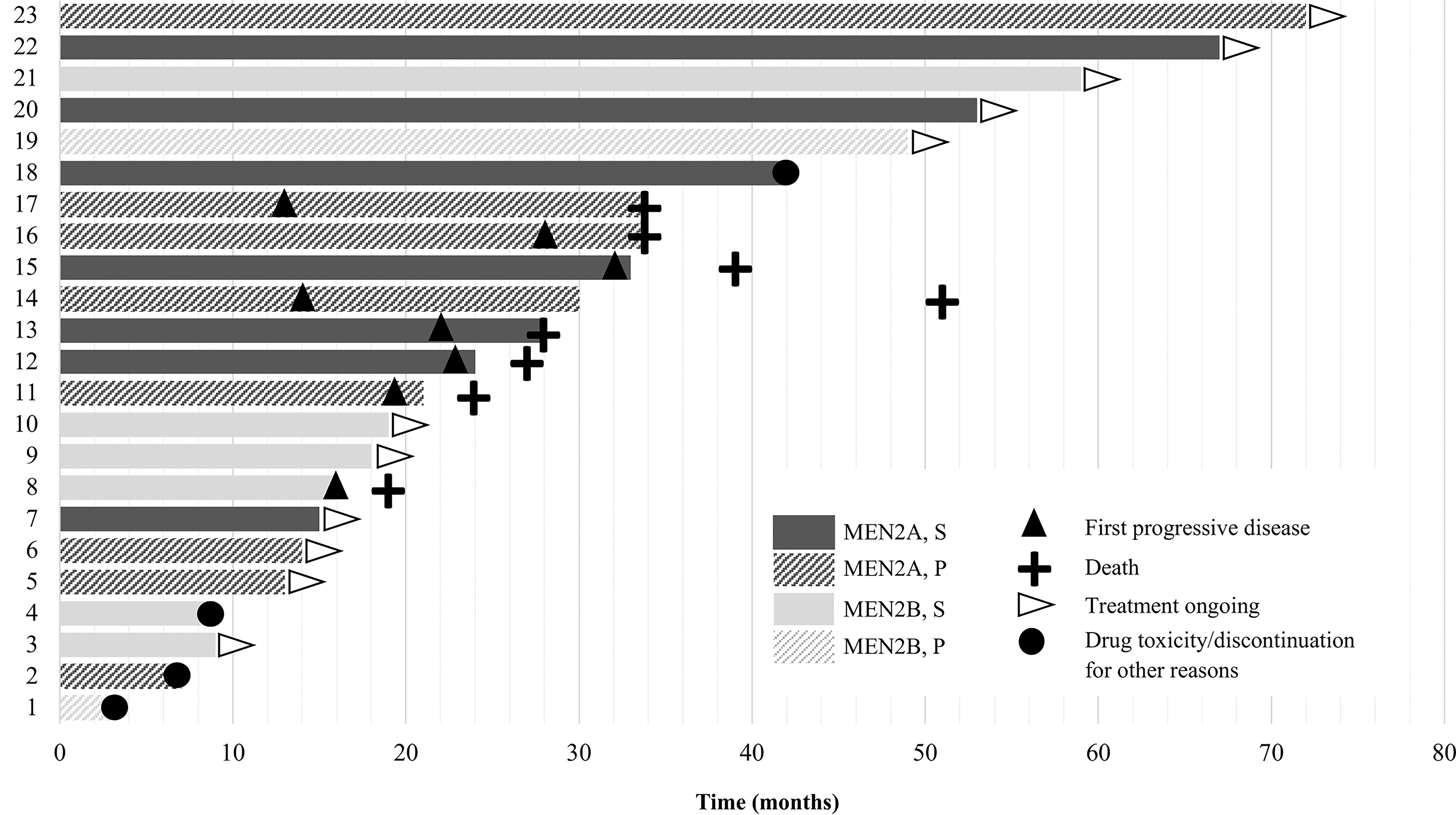
Treatment duration and disposition of 23 patients with advanced hereditary medullary thyroid carcinoma treated with a selective RET inhibitor. S, selpercatinib; P, pralsetinib.
Somatic Mutations Identified at Time of Progression on a Selective RET Inhibitor in Seven Patients with Hereditary Medullary Thyroid Carcinoma
LB, liquid biopsy; NGS, next-generation sequencing.
Clinical Summary of the Eight Hereditary Medullary Thyroid Cancer Patients Treated with a Selective RET Inhibitor who Died
TTx at age 8.
MTC, medullary thyroid carcinoma; AJCC8, American Joint Committee on Cancer Cancer Staging Manual, 8th ed.; NE, not evaluable; Y, yes; N, no; EBRT, external beam radiation therapy; RETi, RET inhibitor; BID, twice daily; LN, lymph node(s); PR, partial response; SD, stable disease; CNS, central nervous system.
Four patients stopped therapy for reasons other than death or disease progression. One patient (patient #1), who started on-label pralsetinib for slowly progressive metastatic disease with worsening diarrhea, stopped therapy after three months due to constipation, weight gain, and increased anxiety related to external stressors. They had SD for almost two years following treatment discontinuation, without the need for additional MTC treatment; tumor markers have been very slowly increasing with a doubling time of over seven years. A second patient (patient #2), who was enrolled on the pralsetinib clinical trial for progressive metastatic MTC with disease in the breast, liver, and bones, stopped therapy after seven months due to significant fatigue. At that time, they had an ongoing response to therapy with SD. Despite being off therapy, disease remained stable for 18 months. They have since transferred care to an outside institution with an unknown disease status. A third patient (patient #18) taking on-label selpercatinib stopped therapy after 42 months following a brain hemorrhage (unrelated to a brain metastasis) while on concomitant long-term anticoagulation for a history of thromboembolic disease. At the time of treatment discontinuation, this patient had a complete radiological response, which is ongoing six months after RETi cessation. Similarly, a drug holiday was undertaken in a 17-year-old patient (patient #4) with a target lesion CR after nine months on therapy and baseline disease that was felt not to warrant systemic therapy (started at another institution). They continue to have an excellent response six months later without any further treatment.
Adverse events
Table 5 details the AEs reported in ≥10% of patients during the treatment period as captured in the electronic medical records. All patients experienced at least one AE of any grade. The most frequent all-grade AEs were increase in alanine aminotransferase (ALT) (48%), QT corrected (QTc) interval prolongation (39%), dry mouth (39%), fatigue (35%), increase in aspartate aminotransferase (26%), hypertension (26%), and hyperphosphatemia (26%). Most AEs were grade 1 or 2. Most frequent grade 3 or 4 AEs were elevations in liver enzymes (13%) and decreases in lymphocyte count (9%). One patient (patient #16) developed grade 3 tumor lysis syndrome within one week of initiation of pralsetinib 100 mg twice daily, which led to a 24-hour hospital admission for intravenous hydration and treatment with rasburicase. Pralsetinib was resumed four days later at a reduced dose of 100 mg daily, and he remained on treatment for 34 months until death. Another patient (patient #18), who had been on selpercatinib for 42 months and was receiving concomitant anticoagulation for a history of pulmonary embolism, presented to his local hospital with confusion and aphasia and was found to have an intraventricular and intraparenchymal brain hemorrhage. He required transient external ventricular drain placement and a brief intensive care unit stay. Selpercatinib was discontinued following this episode. No grade 5 treatment-related AEs were observed. Dose reductions due to AEs occurred in eight (35%) patients. When comparing the prevalence of AEs in our study with the three sMTC-predominant published clinical trials, 16,18,19 there were significantly more ALT elevations and QTc interval prolongations in our cohort of patients with hMTC (Table 5). Nonetheless, all QTc prolongations were grade 1–2 and did not require dose reductions, while only one ALT elevation was grade 4, leading to drug interruption. Dry mouth was also significantly more common in our study compared with the ARROW trial, 18 but similar to the selpercatinib trials. 16,19 Occurrence of all other AEs was similar between our study and the clinical trials.
Adverse Events That Occurred in at Least 10% of Patients with Hereditary Medullary Thyroid Cancer Treated with a Selective RET Inhibitor and Comparison with Published Clinical Trials a
Note that the clinical trials also included patients with RET fusion-positive thyroid cancers.
p-Values provided for comparison of adverse events of all grades. Statistically significant results are bolded.
NR, not reported.
Discussion
MTC, both in its sporadic and hereditary forms, is primarily a RET-driven disease. Currently, there are three approved kinase inhibitors for the treatment of advanced MTC (vandetanib, cabozantinib, and selpercatinib) that all inhibit RET activity to variable degrees. 11 Although pralsetinib was initially granted an accelerated approval from the U.S. Food and Drug Administration for RET-mutated MTC, this was recently withdrawn because the phase III trial that needed to obtain full regulatory approval was not done. Similarly, only selpercatinib is approved by the European Medicines Agency for advanced RET-mutant MTC. The advent of selective targeted therapies against the abnormal RET receptor tyrosine kinase has revolutionized the treatment of patients with RET-altered MTC. 15,25 These agents are not only more effective but also have a more favorable safety profile compared with the multikinase inhibitors vandetanib and cabozantinib, 19 which often cause off-target side effects due to inhibition of VEGFR activity. To the best of our knowledge, this is the first reported series of patients with advanced hMTC treated with a RETi. Our study, which incorporates our real-word experience, confirms that selpercatinib and pralsetinib are not only effective but also safe in hMTC patients, despite the concern that the side effect profile might be worse in patients who have a germline pathogenic variant in RET.
The pivotal phase I/II trials LIBRETTO-001 16,17 and ARROW 15,18 included a small number of patients with hMTC (25/134 in ARROW, unknown in LIBRETTO-001), but neither presented results specifically in this subset of patients. In our study, clinical outcomes were comparable to what has been reported in the clinical trials, as detailed in Table 6. With a median follow-up of 49 months, median OS in our study was 51 months (95% CI, 40.5–61.3), which is also in accordance with the excellent survival outcomes reported in all prior clinical trials. 15,16,19 Although median OS was not reached in these trials, duration of follow-up was longer in our study, which could explain the discrepancy.
Efficacy of Selective RET Inhibitors in RET-Altered Medullary Thyroid Carcinoma in Our Study of Hereditary-Only Disease Compared to Published Clinical Trials
MTC, medullary thyroid cancer; ORR, overall response rate; PFS, progression-free survival; MEN, multiple endocrine neoplasia.
MTC presents at a younger age in hereditary forms of the disease compared with sporadic cases. 1,2,5 While no difference in overall and stage-dependent disease-specific survival has been found between sMTC and hMTC, 2,5,26,27 older age at initial diagnosis has been consistently identified as a negative prognostic factor for MTC. 2,9,26,28 In addition, mutational profiles differ between hMTC and sporadic RET-mutant MTCs, which can also harbor RET alterations other than point mutations, such as indels. 29 Most RET mutations giving rise to MEN2A-associated MTC occur within the cysteine-rich extracellular domain (exons 10 and 11), most frequently p.C634X. 21,30 In contrast, most somatic RET mutations are in exon 16, causing a methionine to threonine substitution within the kinase domain (p.M918T). 11,31 Yet, despite their differences, sMTC and hMTC appear to respond similarly to treatment with RETi, although no direct head-to-head comparison has been made.
Selpercatinib and pralsetinib were also well tolerated among patients with hMTC. Certain conditions associated with MEN2, such as constipation due to Hirschsprung’s disease in MEN2A and intestinal dysmotility in MEN2B, would suggest a potential for increased drug-induced toxicity in this patient population. Nevertheless, the incidence and severity of most AEs in our cohort were comparable to what has previously been reported in clinical trials, which comprised mostly sMTC patients 15 –19 (Table 5). This is in accordance with a recent case series that included five patients with MEN2A-associated pheochromocytoma treated with selpercatinib, in whom no new or unexpected AEs were reported. 32 While all patients experienced at least one treatment-related AE, most were grade 1–2. Hypertension of any grade was reported in 26% of patients in our cohort, compared with 30% and 42% with selpercatinib in the LIBRETTO-001 and LIBRETTO-531 trials, respectively, and 33% with pralsetinib in ARROW. Similarly, constipation of any grade occurred in 17% of our patients with hMTC compared with 16% in LIBRETTO-001 and LIBRETTO-531 and 27% in ARROW. ALT elevation and QTc interval prolongation were identified more commonly in our study, suggesting that the germline nature of the RET pathogenic variant in hMTC may predispose to some increased cardiac and liver toxicity. However, it remains unclear if this is a true difference in toxicity occurrence or if the difference observed is simply due to the smaller sample size in our study compared with the clinical trials. Nevertheless, most of these AEs were grade 1–2, and none led to clinically significant complications, implying that careful monitoring for these toxicities according to the drug labels should be sufficient until more data are obtained.
At progression, molecular testing identified in two tumors acquired RET p.G810X solvent-front mutations, which are known to confer resistance to first-generation RETi. 33 –35 Substitutions of the glycine residue at position 810 with a bulky or charged residue such as cysteine, serine, arginine, or valine directly interfere with drug binding, leading to the development of resistance. 11,36 Interestingly, no secondary RAS pathogenic variant was identified in our cohort, although acquired RAS mutations, which allow bypass activation of the mitogen-activated protein kinase pathway, have been reported in up to 50% of patients who develop resistance to a RETi. 37 Mutations in TP53 and RB1, both tumor suppressor genes, were also observed in 43% of tested tumors, similar to Hadoux and colleagues, who identified TP53 and RB1 alterations in 36% and 14% of tumors at progression on a RETi. 37 Although these mutations do not interfere directly with the mechanism of action of the RETi, they are associated with more aggressive tumor phenotypes, as shown by an increase in the Ki67 index. 37 In one patient, an emergent EML4::ALK fusion was identified. This patient was treated with the combination of alectinib, an ALK inhibitor, and pralsetinib, which only offered a short-lived stabilization of disease. The acquisition of an EML4::ALK fusion has already been described as a potential mechanism of resistance to selpercatinib in another case report. 38 Finally, an FGFR3 p.R471Q missense alteration was identified at progression in one patient, but the contribution of this variant to pralsetinib resistance is unclear as it has not been previously described in thyroid cancer to the best of our knowledge.
In our cohort, four patients stopped the RETi while they had an ongoing response to therapy, all of whom had a sustained response for 6–18 months after drug discontinuation. Thus, it seems that, in selected patients with slowly PD or prolonged sustained responses, drug interruptions to allow improved patient quality of life can be considered, especially given the incurable nature of this disease. This is a particularly interesting observation in patients with hMTC who are often young and could require systemic therapy for a good part of their life. Similarly, a case series has shown that vandetanib discontinuation in patients with MTC after prolonged therapy with documented SD is safe and does not automatically result in rapid disease progression. 39
In conclusion, the selective RETis selpercatinib and pralsetinib are associated with marked and durable responses in patients with advanced hMTC, with a similar toxicity profile to what has previously been described in clinical trials that primarily included sMTC cases. In our cohort, grade 1–2 ALT elevations and QTc interval prolongations occurred more frequently than in the published trials, but none that required a permanent discontinuation of therapy. Further studies are needed to determine whether clinical outcomes, duration of response, and long-term effects of therapy are different in hMTC compared with sporadic cases.
Footnotes
Acknowledgments
The authors acknowledge the research and clinical teams who cared for patients included in this study, and the authors thank the patients and their families for helping them to better understand how to treat advanced hereditary medullary thyroid carcinoma.
Authors’ Contributions
S.H.: Conceptualization, investigation, formal analysis, writing—original draft, review, and editing. S.G.W.: Conceptualization, supervision, provision of patients, review, and editing. S.Y.: Investigation, review, and editing. M.I.H., N.L.B., S.I.S., C.J., E.G.G., A.M., M.E.Z., and V.S.: Provision of patients, review, and editing.
Author Disclosure Statement
N.L.B. has received clinical trial funding from Eisai and Merck. E.G.G. oversees institutional research funding received by MD Anderson from Exelixis and Eli Lilly & Co. M.I.H. has participated in a steering committee and was site PI for Eli Lilly & Co. A.M. has research contracts with Jazz Pharmaceuticals and Thryv Therapeutics Inc. S.I.S. has received clinic trial funding (site PI) from the International Thyroid Oncology group and Eli Lilly & Co. V.S. reports grants/contracts from AbbVie, Inc., Agenus, Amgen, Bayer HealthCare Pharmaceuticals Inc., Blueprint Medicines, Berghealth, Boston Biomedical, Boston Pharmaceuticals, Celgene Corporation, Dragonfly Therapeutics, Exelixis Inc., F. Hoffmann‐La Roche AG, FUJIFILM Medical Systems USA, Inc., Genentech USA, Inc., GlaxoSmithKline, LLC, Idera Pharma, Incyte Corporation, Inhibrx, LOXO Oncology/Eli Lilly, MedImmune, LLC, Multivir, Nanocarrier, the National Comprehensive Cancer Network, the National Cancer Institute, Novartis Pharma, OncLive, Oncusp, PERS, Pfizer, PharmaMar, Relay Therapeutics, and Takeda Oncology; personal/consulting fees from AbbVie, Bayer HealthCare Pharmaceuticals Inc., F. Hoffmann‐La Roche AG, Illumina, Incyte Corporation, Jazz Pharmaceuticals Inc., LabCorp (aka Lab Corporation), Regeneron Pharmaceuticals, Inc., and Relay Therapeutics; support for other professional activities from Aadi Biosciences, Clinical Care Communications, Illumina, Jazz Pharmaceuticals Inc., Med Learning Group, Medscape, and Pheon Therapeutics; and travel support from the American Society of Clinical Oncology and the European Society of Medical Oncology. M.E.Z. has received clinical trial research funding (PI) from Eli Lilly and Merck. All others have no conflicts of interest to disclose.
Funding Information
No funding was received for this work.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2