
Abstract
Background:
Thyroid eye disease (TED) is an autoimmune disorder characterized by proptosis, inflammation, and fibrosis. Elevated insulin-like growth factor 1 receptor (IGF1R) signaling in TED orbital fibroblasts (OFs) drives the proliferation and biosynthesis of hyaluronan, which causes enlargement of orbital tissue volume. The aryl hydrocarbon receptor (AHR) is a ligand-activated transcription factor that regulates cellular stress responses, metabolism, and inflammation. Given its important role in regulating cellular responses, we hypothesized that activation of the AHR could limit excessive IGF1R signaling in TED OFs, offering therapeutic potential.
Methods:
We measured IGF1R and AHR expression levels in TED, non-TED, and non-OF controls. OF activation was analyzed using proliferation, hyaluronan accumulation, and migration assays. RNA sequencing was used to detect transcriptome-wide changes in IGF1-treated TED OFs. After gene set enrichment analysis, select gene expression changes were validated by quantitative polymerase chain reaction. OFs were treated with the AHR ligands 6-formylindolo[3,2-b]carbazole (FICZ) or tapinarof with or without IGF1. Western blotting evaluated signaling pathways impacted by AHR and IGF1R signaling.
Results:
TED OFs showed elevated IGF1R and AHR expression levels compared to controls. IGF1 significantly increased hyaluronan accumulation, proliferation, and migration in TED OFs compared to non-TED OFs. IGF1R signaling altered the expression of hundreds of genes controlling cell migration, proliferation, and metabolism in TED OFs. These genes included TUBA1B, TUBA1C, CRABP2 (upregulated), and IRS2 and SOD3 (downregulated). AHR activation blocked proliferation, migration, hyaluronan production, and gene expression mediated through IGF1R signaling. The AHR inhibited these pathways by reducing phosphorylation of GSK3β, an important mediator of IGF1R/β-catenin mediated signaling.
Conclusions:
AHR activation represents a promising therapeutic strategy for mitigating TED progression by inhibiting IGF1R signaling. Through modulation of GSK3β-mediated pathways, AHR activation may target additional pathologically relevant pathways beyond those affected by direct IGF1R inhibitors. This research provides novel insights into TED pathophysiology and offers a potential avenue for developing therapies to improve patient outcomes.
Introduction
Thyroid eye disease (TED) is a debilitating autoimmune disorder characterized by a range of ocular manifestations, including eyelid retraction, proptosis (bulging eyes), diplopia (double vision), and in severe cases, vision loss. 1 –3 TED affects 25–50% of patients with Graves’ disease, as well as a smaller proportion of individuals with Hashimoto’s thyroiditis or normal t9hyroid function. 4,5 The underlying pathogenesis of this complex disease remains poorly understood. The risk factors for developing TED, such as older age, selenium deficiency, and cigarette smoking, further underscore the multifactorial nature of this disorder. 1
Elevated insulin-like growth factor 1 receptor (IGF1R) signaling is observed in TED, opening important opportunities for targeted interventions. 6 –8 IGF1R signaling promotes cell proliferation and biosynthesis of the glycosaminoglycan, hyaluronan in the orbits of afflicted individuals. 9 Hyaluronan, which can bind up to 1000 times its weight in water, increases orbital volume and swelling through its extreme hydrophilicity. 10 Furthermore, hyaluronan accumulation increases immune cell retention and promotes inflammatory signaling in the orbit. 11,12 The pivotal role of IGF1R is further strengthened by the clinical success of teprotumumab, the first and only TED-specific FDA-approved therapy. 13,14 Teprotumumab is an IGF1R-blocking monoclonal antibody that reduces proptosis, diplopia, and clinical activity scores in 70–90% of TED patients. 14 –16 While teprotumumab has been a significant advance, it is not without shortcomings. For example, 10–30% of patients do not respond to teprotumumab therapy, and some patients experience serious side effects such as hearing loss and muscle spasms, and reported symptom recurrence after completing the six-month course of intravenous injections. 17 –19 Given these limitations, there is a pressing need to identify additional therapeutic targets and treatment strategies that could benefit non-responders or provide more durable responses in TED patients.
In TED, IGF1R is upregulated in TED orbital fibroblasts (OFs), fibrocytes, and T cells. 6,20 In addition to its role in proliferation and hyaluronan production, elevated IGF1R activity may play a role in promoting inflammation, orbital adipogenesis and myofibroblast formation. 21,22 Mechanistically, IGF1R, a receptor tyrosine kinase, drives these pathological changes in part through activation of AKT, 23 –25 which inhibits glycogen synthase kinase 3β (GSK3β) through phosphorylation, 26 leading to increased β-catenin signaling and downstream transcriptional changes. 27 IGF1R may also interact and/or colocalize with the thyrotropin receptor (TSHR), the primary autoantigen in Graves’ disease, and TED 23 –25 to mediate pathological changes.
The aryl hydrocarbon receptor (AHR) is a ligand-activated transcription factor that exhibits complex and context-dependent roles in homeostasis and disease, including TED pathophysiology. 28,29 AHR responds to diverse ligands including toxins like dioxin and cigarette smoke components, 28,29 and endogenous compounds such as tryptophan derivatives like 6-formylindolo[3,2-b]carbazole (FICZ) and microbiome metabolites such as indole-3 acetic acid. 30 Previous studies revealed that AHR activation by FICZ blocks the fibrotic response driven by transforming growth factor-β (TGFβ) in TED OFs. 31,32 Proton-pump inhibitors, such as esomeprazole, also activate the AHR to limit TGFβ signaling in TED OFs. 33 Tapinarof is a naturally derived AHR ligand that resolves topical inflammation in both mice and humans. 34 The therapeutic potential of AHR modulation is supported by the clinical success of tapinarof in treating plaque psoriasis. 35,36 AHR activation can enhance GSK3β activity by reducing its inhibitory phosphorylation, 31,33,37 suggesting potential crosstalk between AHR and IGF1R signaling pathways that could be therapeutically relevant in TED.
The AHR and IGF1R pathways are both poised as central mediators in TED-relevant pathways of fibroblast activation, migration, and hyaluronan production. Currently, it is unclear whether there is crosstalk between the IGF1R and AHR pathways. The aim of this study is to elucidate the interplay between the IGF1R and AHR. We hypothesize that AHR offers therapeutic potential by limiting IGF1R signaling in TED. Here, we investigate the expression and functional interactions of IGF1R and AHR in TED and identify novel target genes and pathways that mediate IGF1-dependent TED OF proliferation, migration, and hyaluronan production.
Methods
Sample selection, fibroblast isolation, and cell culture
The study was conducted following the tenets of the Declaration of Helsinki as revised in 2024. All procedures were conducted in accordance with ethical standards and approved protocols. This study was approved by the University of Rochester Medical Center (URMC) Research Subjects Review Board (RSRB, approval #00023924). Orbital tissue explants were collected during surgical decompression for TED and other non-TED eye surgeries at the Flaum Eye Institute, URMC, with informed consent from all patients. TED patients included in the study had clinical indications for decompression surgery due to severe orbital disease. The decompression surgeries were performed for functional recovery and were not cosmetic in nature. The clinical severity of the disease required these surgical interventions to alleviate symptoms and improve patient outcomes. Non-TED, control tissue: Normal orbital tissue was obtained as scrap tissue from patients undergoing bone fracture repairs. These patients had medical issues necessitating orbitotomy that were unrelated to any orbital pathology. The tissue was collected as part of routine surgical procedures and would otherwise be discarded. This approach ensured that the control tissue was free from any disease-related alterations. All consecutively available tissue samples were acquired and used for this study. This selection method helped minimize selection bias and ensured that the samples were representative of the patient population undergoing these procedures. Relevant patient demographic data is listed in Table 1. Subcutaneous abdominal fat tissue explants from anonymized surgical specimens were obtained from the Pathology department at the URMC. Subcutaneous fat samples were deidentified and determined to be exempt by the RSRB from the requirement for informed consent. Fibroblasts were grown out of tissue as described previously. 31,32 IGF1 was obtained from PeproTech (Cranbury, NJ) and used at 10–50 ng/mL. 0.1% BSA-PBS served as the vehicle for IGF1. The AHR ligands FICZ (Enzo Life Sciences, Farmingdale, NY) and tapinarof (Selleckchem, Houston, TX) were dissolved in DMSO (vehicle) (Sigma-Aldrich, St. Louis, MO).
Patient Demographic Summary
TED, thyroid eye disease.
Western blotting
Protein lysates were prepared as previously described 32 and probed with the antibodies listed in Supplementary Data. Densitometry was performed using Bio-Rad Image Lab Software.
Scratch/migration assay
The migratory capacity of OFs was assessed using the scratch assay. 38 A sterile pipette tip was used to create a vertical scratch approximately 1 mm wide across a confluent cell monolayer. Images of the scratch area were captured at 0, 24, 48, and 72 hours posttreatment. The degree of wound closure was quantified using ImageJ (National Institutes of Health, Bethesda, Maryland). The open area of the scratch was outlined and measured at each time point and the area at time 0 was set to 1.0.
Cell proliferation assay
Cell proliferation was determined using the bromodeoxyuridine (BrdU) Cell Proliferation Assay kit (Calbiochem, San Diego, CA) as described previously. 39 OFs were seeded in a 96-well plate at a density of 5 × 103 cells/well and treated as indicated.
Hyaluronan detection
Culture supernatants were collected and analyzed for hyaluronan (HA) levels using the HA DuoSet ELISA from R&D Systems (Minneapolis, MN), following the manufacturer’s instructions. Some fractions of supernatants were treated with hyaluronidase (Sigma-Aldrich) to ensure the specificity of the assay. 12
Gene expression knockdown using siRNA
Silencer™ Select Pre-Designed siRNA for AHR (4390824, ID number: s1199) and a nonspecific Silencer™ Select Negative Control No. 1 (4398044) were purchased from ThermoFisher Scientific. Cells were treated with siRNAs (50 nM) mixed with RNAiMAX Transfection Reagent in Opti-MEM™ I Medium (Invitrogen, Carlsbad, CA) for 48 hours. Cells were then treated with or without IGF1, FICZ, or tapinarof as shown.
RNA isolation and quantitative polymerase chain reaction
Total cell RNA was extracted and quantified as described previously. 32 Real-time PCR with the gene-specific primers (Integrated DNA Technologies, Coralville, Iowa) listed in Supplemental Data was performed as described previously. 32
RNA-seq library preparation and sequencing
Total RNA was isolated and processed by the University of Rochester’s Genomics Research Center (Rochester, NY). Total RNA was extracted using the QIAGEN RNeasy mini kit (Qiagen Sciences, Germantown, MD), assessed via an Agilent Bioanalyzer (Agilent Technologies, Santa Clara, CA), and prepared for analysis via a TruSeq Stranded mRNA Sample Preparation Kit (Illumina, Inc., San Diego, CA). Single end-reads of 50 nucleotides were generated for each sample using Illumina’s NovaSeq6000 (San Diego, CA).
Bioinformatics analysis
The data for this project was deposited to NCBI with the following accession: GSE276265. Differentially expressed genes were identified (DESeq2) between the control and IGF1-stimulated groups. Gene lists were submitted to the DAVID Bioinformatics Resources (https://david.ncifcrf.gov/) for functional annotation. The “Homo sapiens” background gene list was used for comparison. Statistical significance was determined using a Benjamini–Hochberg adjusted p value.
Statistical analysis
Statistical analyses were performed using GraphPad Prism v8.0. All data were reported as mean ± standard error of the mean. Specific statistical tests employed depended on the experimental design and the number of groups being compared. For data from two groups, unpaired Student’s t-tests were used. For data involving three or more groups, a one-way analysis of variance (ANOVA) was performed. Post hoc Bonferroni’s multiple comparisons tests were then utilized to identify specific group differences. For comparing treatment and time in the scratch migration assays, two-way ANOVA, followed by Dunnett’s posttest was performed. Sample sizes were chosen based on previous studies and power calculations to ensure sufficient statistical power for detecting treatment effects. Sample sizes ranged from N = 3 to N = 5, depending on the specific assay.
Results
Elevated IGF1R and AHR expression in TED orbital fibroblasts
OFs derived from TED and non-TED patients as well as fibroblasts derived from a non-ocular tissue (subcutaneous abdominal fat-derived fibroblasts (SubQ) were cultured with or without the IGF1R ligand, IGF1 (50 ng/ml), for 48 hours. Cells were then lysed and analyzed by Western blotting for IGF1R, AHR, and actin (Fig. 1). The antibody for IGF1R recognized both the pro-form of the protein (upper band) and the mature, active form of the receptor (lower band). TED OFs had ∼3.5-fold higher levels of the mature form of IGF1R compared to SubQ fibroblasts (Fig. 1A). TED OFs also showed ∼5-fold more AHR expression levels than SubQ fibroblasts. TED OFs further displayed elevated levels of both IGF1R (∼2-fold) and AHR (∼1.8-fold) compared to non-TED OFs (Fig. 1B). We were also able to confirm AHR expression in primary TED orbital tissue using immunohistochemistry (Supplementary Fig. S1).
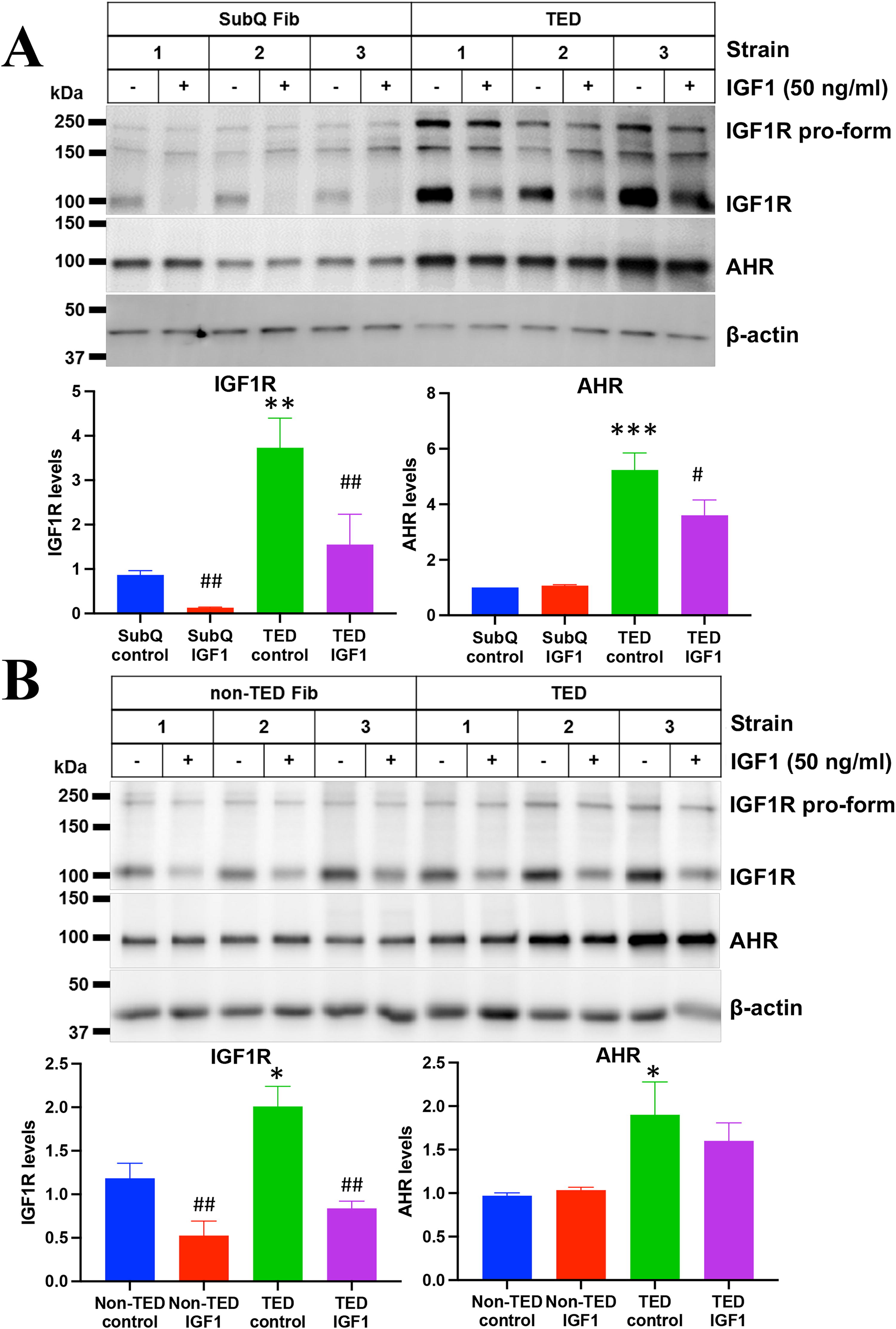
Elevated IGF1R and AHR expression in TED orbital fibroblasts. Orbital fibroblasts from patients with thyroid eye disease (TED) exhibit significantly higher basal levels of IGF1 receptor (IGF1R) and aryl hydrocarbon receptor (AHR) compared to both subcutaneous fat-derived fibroblasts (SubQ)
AHR activation inhibits IGF1-induced cellular migration and proliferation
To assess the impact of AHR activation on IGF1R-dependent cell migration and proliferation, both migration and BrdU incorporation assays were performed on TED and non-TED OFs (Fig. 2). For the migration assay, confluent cell layers were scratched as described in the Methods section. Cells were treated with or without IGF1 (50 ng/mL) and the AHR ligand FICZ (1 μM) for 72 hours. Visual inspection revealed migration patterns of treated and control cultures (Fig. 2A). IGF1 treatment significantly increased the migration of TED OFs compared to non-TED OFs (Fig. 2B). At 72 hours after IGF1 treatment, TED OFs had migrated over the open areas to leave only 40% uncovered regions while, in non-TED OFs, around 60% of the open regions remained. In both TED and non-TED OFs, FICZ significantly attenuated IGF1-induced cell migration. The effect was more pronounced in TED OFs (∼2-fold attenuation) than in non-TED OFs (∼1.5-fold). FICZ also prevented baseline migration in OFs.
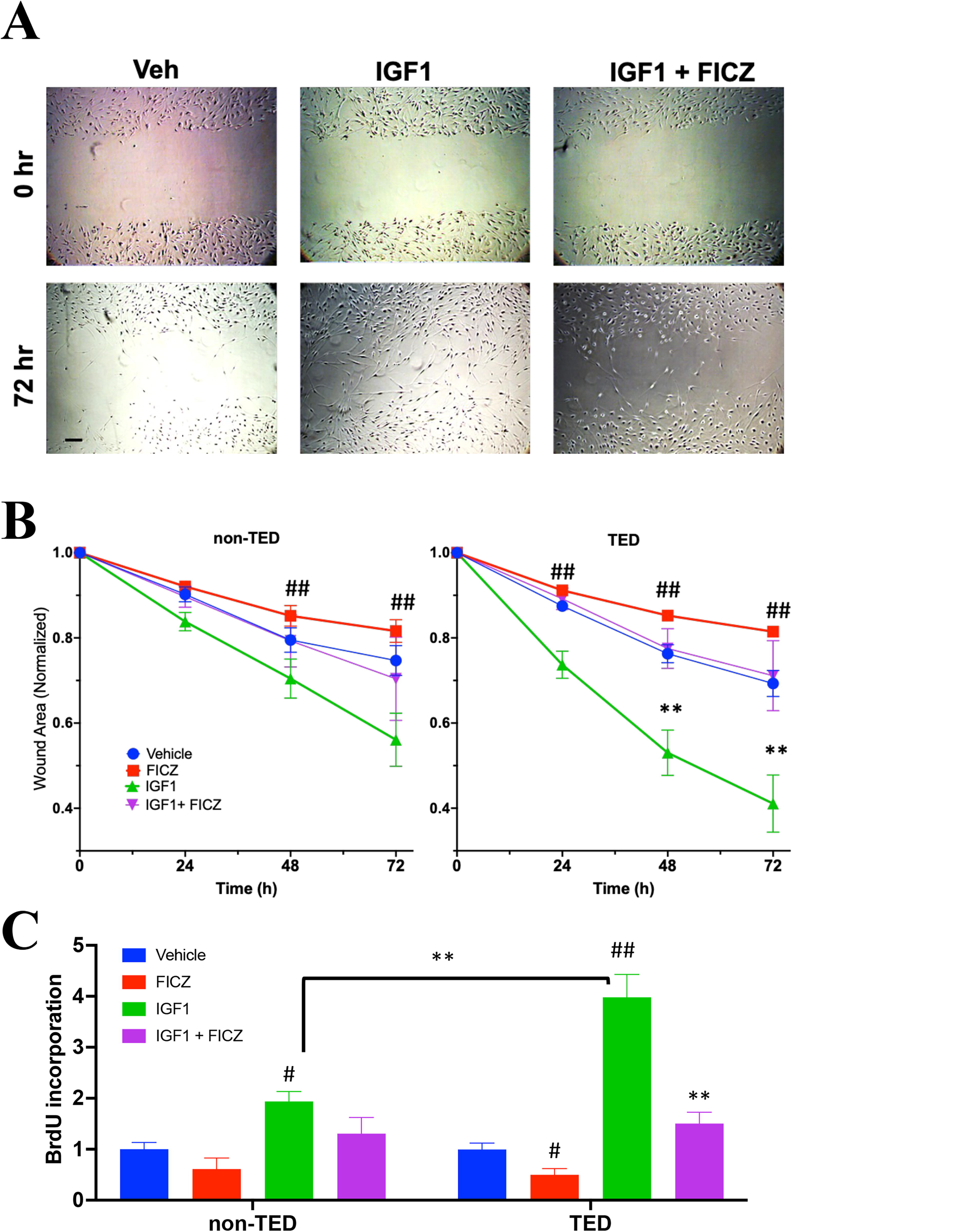
AHR activation reduces IGF1-induced TED orbital fibroblast migration. Orbital fibroblasts from TED and non-TED patients (N = 4 from each type) were grown to a confluent monolayer. After scratching the cell layer, cells were treated with IGF1 (50 ng/mL) or IGF1 plus FICZ (1 µM) for 72 hours to assess migration. Areas without cell migration were quantified using ImageJ Software and normalized to the initial scratch (defined as 1.0 at time 0). Images were captured at 0, 24, 48, and 72 hours.
The BrdU incorporation assay was used to analyze cell proliferation rates. Here, non-TED and TED OFs were seeded into 96 well plates and treated as above. During the incubation, the thymidine analog, BrdU, was added to measure relative proliferation levels (Fig. 2C). Upon IGF1 treatment, TED OFs incorporated 2-fold more BrdU than non-TED OFs, indicating higher proliferation rates and greater IGF1 responses in TED OFs. FICZ treatment reduced both basal and IGF1-induced proliferation in TED OFs. These results show that IGF1 increases both migration and proliferation in TED OFs and that ligand-mediated activation of the AHR attenuates these effects.
AHR activation inhibits IGF1-induced hyaluronan accumulation
Hyaluronan is a critical regulator of TED pathophysiology. In addition to its excessive accumulation in the orbits of TED patients, which causes swelling and increases in tissue volume, hyaluronan actively participates in cell differentiation, proliferation, and immune cell trafficking. 10,12,40 In TED OFs, hyaluronan biosynthesis is driven in part through IGF1R. 6,40 To further study the role of IGF1R and AHR signaling in OFs, both non-TED and TED OFs were treated with or without IGF1 (50 ng/ml) and FICZ (1 μM) for 72 hours. Afterward, culture supernatants were isolated and analyzed for hyaluronan as described in the Methods (Fig. 3). Both non-TED and TED OFs increased hyaluronan synthesis in response to IGF1. TED OFs generated around 70% more hyaluronan than non-TED OFs. AHR activation through FICZ blocked IGF1-induced hyaluronan accumulation in both non-TED and TED OFs.

IGF1 induces hyaluronan (HA) production in non-TED and TED which is blocked by the AHR ligand, FICZ. TED and non-TED orbital fibroblasts (N = 4 from each type) were treated with IGF1 (50 ng/mL), FICZ (1 µM) or IGF1 plus FICZ for 72 hours. After treatment, the culture supernatant was isolated and analyzed for HA accumulation by an ELISA-like assay as described in the Methods section. IGF1 significantly induced HA production in both non-TED (gray bars) and TED OFs (black bars). IGF1 increased HA significantly more in TED OFs (***p < 0.001) than in non-TED OFs. FICZ significantly blocked IGF1-induced (****p < 0.0001) hyaluronan in TED OFs.
Transcriptome analysis reveals IGF1-induced gene expression changes in TED orbital fibroblasts
To better understand the downstream effects of IGF1 signaling in TED, RNA sequencing was performed on two different TED OF strains treated with or without IGF1 (50 ng/ml) for 24 hours. Heatmap analysis (Fig. 4A) highlighted distinct clusters of genes upregulated (red) and downregulated (blue) by IGF1. A volcano plot (Fig. 4B) further confirmed broad expression changes. Venn diagrams (Fig. 4C and D) revealed considerable overlap of differentially expressed genes (DEGs) between the two strains. Specifically, 95 genes were upregulated at least 2-fold, and 255 genes were downregulated at least 0.5-fold in both strains.

Transcriptome analysis reveals IGF1-induced gene expression changes in TED orbital fibroblasts. Two strains of TED orbital fibroblasts were treated with IGF1 (50 ng/mL) for 24 hours and analyzed by RNA sequencing (N = 3 samples/group). Differential expression analysis using DAVID identified significantly upregulated and downregulated genes.
Common DEGs were analyzed by gene ontology (GO) enrichment using DAVID. 41,42 Upregulated genes were enriched for several GO processes including microtubule cytoskeleton components, biosynthetic/metabolic processes, and the cell cycle (Table 2). For example, TUBA1B and TUBA1C were significantly induced by IGF1 in TED OFs. These genes encode α-tubulin chain components that promote the cell cycle, cytoskeleton-dependent transport, and cellular migration. 43 The hyaluronan synthase gene, HAS2 was upregulated by IGF1 in TED OFs. Other genes encoding proteins involved in metabolism and lipid transport, such as the fatty acid binding proteins, FABP3 and FABP5, and cellular retinoic acid binding protein 2 (CRABP2) were also upregulated. Interestingly, CRABP2 expression has been shown to increase PI3K/Akt signaling which blocks GSK3β signaling to promote cell migration, suggesting another potential mechanism whereby IGF1R signaling can promote TED pathogenesis. 44
Gene Ontology Enriched Terms in the Gene List of IGF1 Upregulated Genes by DAVID Analysis
Fold enrichment represents the ratio of genes enriched in the gene list compared to the background gene list. p Values are adjusted for multiple comparisons by the Benjamini–Hochberg false discovery rate (FDR) correction.
GO, gene ontology.
While IGF1R signaling induced the expression of many genes in TED OFs, a larger portion of genes was significantly repressed by IGF1 treatment. GO analysis revealed that pathways enriched in the IGF1 downregulated genes included adhesion, migration, chromatin remodeling, and response to hypoxia (Table 3). Insulin receptor substrate-2 (IRS2), a cytoplasmic protein that mediates both insulin and IGF1 signaling was significantly reduced in IGF1 treated OFs. IRS2 is an adaptor protein that links receptor tyrosine kinases with other signaling proteins. 45 Superoxide dismutase 3 (SOD3), an enzyme involved in mitigating oxidative stress, inflammation, and cell migration, 46,47 was reduced by IGF1. These results highlight the effects of IGF1 signaling on gene expression and cell activation in TED OFs.
Gene Ontology Enriched Terms in the Gene List of IGF1 Downregulated Genes by DAVID Analysis
Fold enrichment represents the ratio of genes enriched in the gene list compared to the background gene list. p Values are adjusted for multiple comparisons by the Benjamini–Hochberg false discovery rate (FDR) correction.
AHR activation mitigates IGF1 signaling
RT-quantitative polymerase chain reaction (qPCR) was used to confirm IGF1-induced gene expression changes obtained from RNA-seq. Some cells were also treated with FICZ, with or without IGF1. IGF1 induced the mRNA levels of TUBA1B (∼1.8-fold), TUBA1C (∼ 2-fold), and CRABP2 (∼2.3-fold), supporting the RNA-seq findings (Fig. 5A). FICZ treatment significantly reversed this effect on gene expression. Interestingly, FICZ treatment alone significantly reduced the expression of both TUBA1B and CRABP2, suggesting that AHR activation can alter these pathways independently of IGF1R signaling. RT-qPCR also confirmed that IGF1 reduced SOD3 (∼70% down) and IRS2 (∼25% down) mRNA in TED OFs (Fig. 5B). FICZ restored and even induced these genes compared to vehicle samples. Interestingly, FICZ treatment increased the expression of IRS2 and SOD3 mRNA levels without IGF1. These results demonstrate that FICZ mitigates the effects of IGF1 on gene expression in TED OFs and suggests that the AHR and IGF1R pathways crosstalk in TED OFs. The AHR target gene, CYP1B1, was used to confirm AHR-dependent signaling (Fig. 5C).
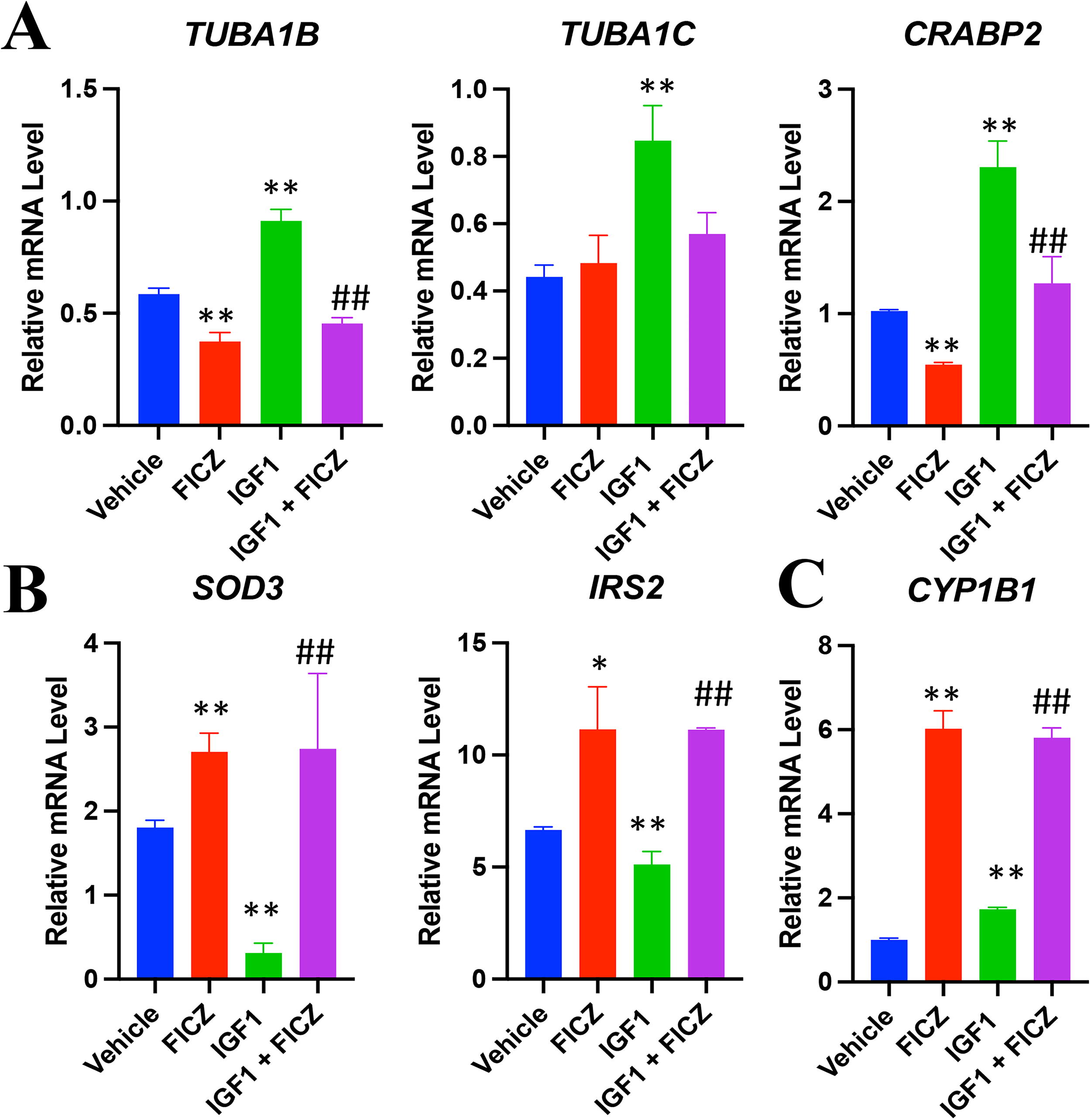
AHR activation mitigates IGF1 signaling. TED orbital fibroblasts were treated with vehicle, FICZ (1 µM), IGF1 (50 ng/mL), or IGF1 plus FICZ (1 µM) for 24 hours. After treatment, cells were isolated and RNA levels were measured by RT-qPCR.
AHR blocks IGF1-induced protein expression changes and modulates GSK3β signaling
To further explore AHR and IGF1R crosstalk in TED OFs, relevant protein expression and phosphorylation status were analyzed (Fig. 6). CRABP2 displayed a dose-dependent upregulation with IGF1 treatment, where 10 ng/mL IGF1 induced CRABP2 protein levels by ∼2.7-fold and 50 ng/mL IGF1 induced CRABP2 by ∼4 fold (Fig. 6A). The addition of FICZ significantly blocked CRABP2 expression. FICZ also restored and even induced SOD3 levels in control and IGF1-treated samples.
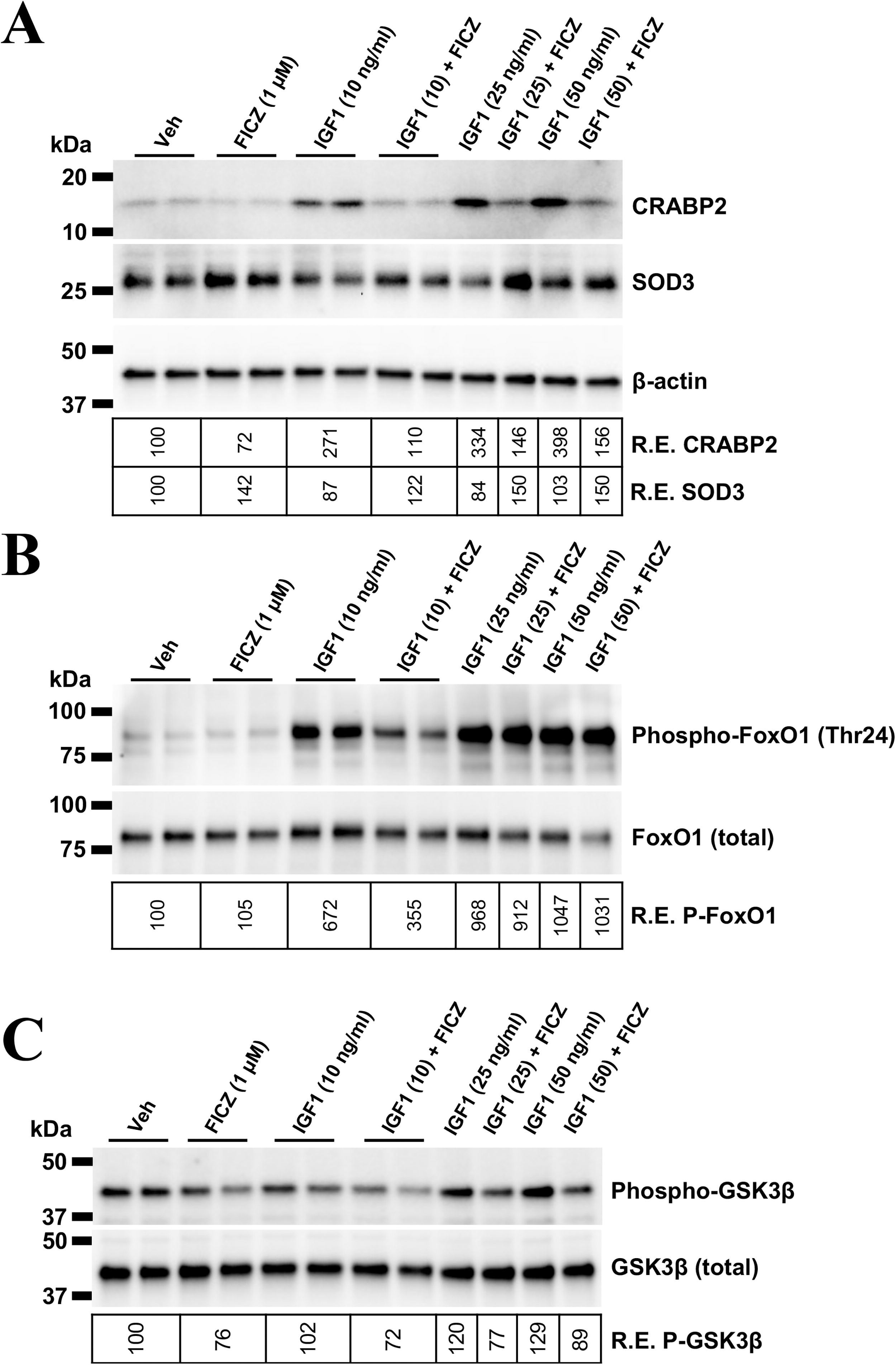
AHR activation mitigates IGF1-induced CRABP2 expression, SOD3 downregulation, and FOXO/GSK3β phosphorylation in TED orbital fibroblasts. TED orbital fibroblasts were treated with vehicle, the AHR ligand, FICZ (1 µM), or varying doses of IGF1 with or without FICZ for 48 hours. Protein levels were analyzed by Western blotting.
To further examine the mechanism(s) driving AHR-dependent antagonism of IGF1R signaling, downstream IGF1R targets FOXO and GSK3β were studied. 48,49 FOXO1 is a Forkhead box O transcription factor involved in proliferation, senescence, and metabolism. 49,50 Upon IGF1R activation, FOXO1 undergoes inhibitory phosphorylation at Ser256. To test if AHR alters IGF1R signaling through FOXO1 activity, phospho-FOXO1 was analyzed (Fig. 6B). IGF1 treatment induced a 6-to-10-fold induction of phospho-FOXO1. FICZ attenuated FOXO1 phosphorylation at the lowest IGF1 dose (10 ng/mL) by ∼50%. However, FICZ lost its efficacy at higher IGF1 levels (25 and 50 ng/mL). IGF1 increased GSK3β phosphorylation at Ser9 (Fig. 6C). Both basal and IGF1-induced GSK3β phosphorylation was blocked by FICZ, suggesting that GSK3β may be a key factor in AHR-IGF1R pathway crosstalk.
AHR knockdown increases IGF1-induced GS3K3β phosphorylation and hyaluronan production
Since AHR activation could block IGF1-induced cell migration, cell proliferation, and HA production, we examined if AHR depletion could alter IGF1-dependent signaling. To test this, control or AHR-specific siRNAs were introduced into TED OFs. Cells were treated with or without IGF1 (50 ng/ml), FICZ (1 μM), or tapinarof (1 μM) for 72 hours. To confirm AHR knockdown and measure IGF1R signaling through GSK3β, OFs were isolated and analyzed by Western blot (Fig. 7A). In control siRNA-treated samples, IGF1 treatment increased GSK3β phosphorylation by ∼150%. FICZ and tapinarof were able to block GSK3β phosphorylation in both basal and IGF1-induced conditions. AHR siRNA reduced AHR expression by around 60% compared to control. Inhibitory GSK3β phosphorylation levels were elevated in AHR siRNA samples compared to control suggesting that AHR promotes GSK3β activity. Furthermore, the ability of FICZ or tapinarof to block GSK3β phosphorylation was markedly lower in AHR siRNA samples.

Depletion of AHR increases IGF1-induced GSK3β phosphorylation and hyaluronan (HA) production. TED orbital fibroblasts were cultured to a confluent layer and transfected with control or AHR siRNA using RNAiMAX lipofection 48 hours before treatment with IGF1(50 ng/mL) and/or the AHR ligands, FICZ or tapinarof (each at 1 μM) for 72 hours.
We next tested if depletion of AHR promotes hyaluronan production in TED OFs (Fig. 7B). In control siRNA samples, IGF1 stimulation significantly increased hyaluronan production compared to vehicle (gray bars). Both FICZ and tapinarof blocked hyaluronan production in control siRNA, IGF1 treated samples. AHR knockdown increased hyaluronan production in response to IGF1R activation (black bars). In the presence of AHR siRNA, both FICZ and tapinarof lost the ability to reduce hyaluronan accumulation. These findings reveal a critical role for AHR in reducing IGF1R-induced GSK3β phosphorylation and hyaluronan accumulation (Fig. 8).
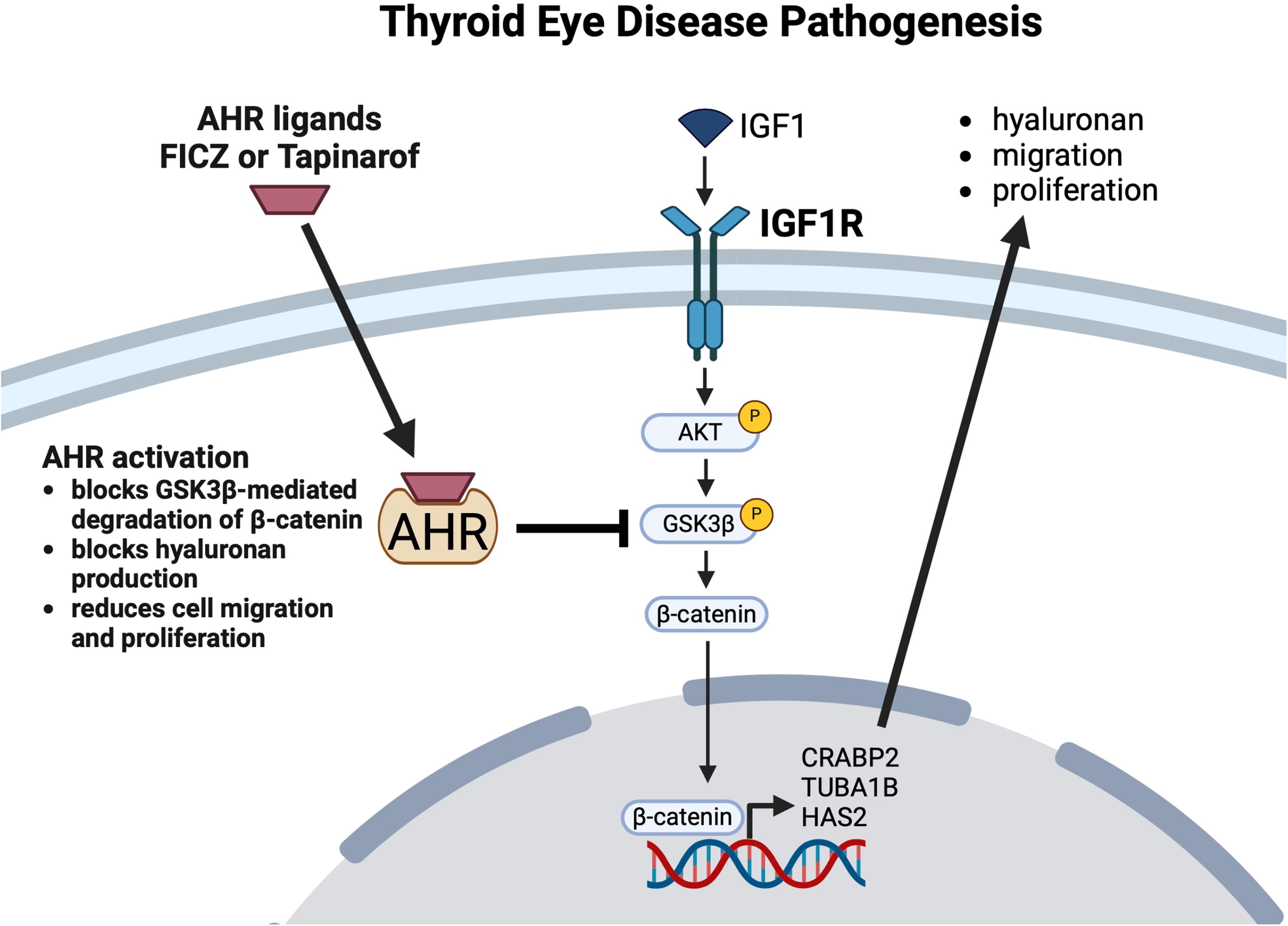
A model illustrating the interactions between IGF1R, AHR, and GSK3β signaling pathways in TED. AHR prevents GSK3β phosphorylation to limit IGF1R induced β-catenin signaling, cell migration, proliferation, and hyaluronan accumulation. IGF1 signaling activates IGF1R, which in turn stimulates the phosphorylation of AKT. Activated AKT phosphorylates GSK3β, inactivating it and preventing the degradation of β-catenin. Stabilized β-catenin promotes cellular processes such as proliferation, migration, and hyaluronan production, in part through activation of genes like CRABP2, TUBA1B, and HAS2, contributing to TED pathogenesis. However, activation of the AHR by FICZ or tapinarof blocks the phosphorylation of GSK3β, allowing the β-catenin destruction complex to remain active in the cytoplasm. This leads to enhanced β-catenin degradation and suppression of its downstream transcriptional activity. As a result, AHR activation reduces cell proliferation, migration, and hyaluronan production, highlighting its potential therapeutic role in TED.
Discussion
We demonstrate that TED OFs show higher IGF1R expression compared to non-TED OFs and subcutaneous fibroblasts. OFs from TED patients proliferate at a higher rate, migrate more rapidly, and produce more hyaluronan than non-TED OFs. We also observe that IGF1 signaling drives changes in gene expression related to these pathways in TED OFs. Further, the AHR ligands, FICZ, and tapinarof block IGF1-induced signaling and hyaluronan accumulation in TED OFs. These data highlight a novel crosstalk between AHR and IGF1R signaling in TED.
Despite the valuable insights gained from this study, some limitations should be acknowledged. Our investigation focused on fibroblasts, which are known to play a central role in TED pathogenesis. However, other cell types, including fibrocytes, T cells, and B cells, contribute to the complex immunopathological processes in TED. 1 Future studies should explore the interplay between IGF1R-AHR signaling and other cell types implicated in TED to obtain a more comprehensive understanding of the disease mechanisms.
Previous studies revealed that T cells, B cells, and fibroblasts from TED patients express high levels of IGF1R. 20,51 Here, we confirm these findings and show that IGF1R is elevated in TED OFs compared to either non-TED OFs or fibroblasts derived from subcutaneous fat depots. However, even non-TED OFs show higher levels of IGF1R than subcutaneous fat fibroblasts. Additionally, IGF1R further supports elevated hyaluronan biosynthesis in TED OFs. These findings suggest that the orbit is poised for elevated IGF1R signaling and hyaluronan accumulation compared to other tissues and may partially explain why the orbit is the main tissue targeted in TED. These data provide support that fibroblasts display tissue-specific phenotypes consistent with previous findings. 52,53
The interplay between TSHR/IGF1R and AHR signaling pathways warrants consideration in the context of TED. The interaction between TSHR and IGF1R has been implicated in the pathogenesis of TED, with TSHR activating IGF1R and promoting proliferation and extracellular matrix production. 23,40 Our study provides evidence that AHR signaling interferes with IGF1R signaling, potentially acting as a negative regulator of TSHR/IGF1R-mediated effects. This raises the possibility that AHR activation could modulate the TSHR/IGF1R axis, attenuating its pathological consequences in TED.
GSK3β is a serine/threonine kinase that regulates various cellular processes, including metabolism, cell cycle, and apoptosis. GSK3β is most well known as mediating wnt signaling, however, wnt-independent changes in GSK3β activity are being increasingly recognized as important in fibrosis and cell proliferation. 54 The most well-studied role for GSK3β in both wnt-dependent and independent signaling is targeting β-catenin for proteasomal degradation. β-catenin, a pro-fibrotic transcription factor, regulates proliferation, 55 migration, 56 and hyaluronan production 57 in cancer and other tissues. For example, in thyrocytes, evidence shows that both TSH and IGF1 induce β-catenin and cell proliferation. 58 Previous studies have highlighted the interplay between IGF1R, GSK3β, and β-catenin, 26,27 yet to our knowledge, this pathway has not been investigated in TED. Our results here suggest a key role for this pathway in TED pathophysiology. Here, we show that AHR promotes GSK3β activity by blocking its phosphorylation in both untreated and IGF1-treated states (Fig. 8). Therefore, AHR activity leads to a loss of β-catenin activity. This provides a key mechanism linking AHR activation and limiting TED pathogenesis. Previous work has revealed a link between AHR and GSK3β signaling 31,37 ; however, more study is needed to elucidate this pathway in TED. Taken together, these results suggest that AHR activation can activate GSK3β by preventing its phosphorylation. This would suggest that AHR activation would promote β-catenin degradation and loss of β-catenin/TCF transcriptional activity.
AHR ligands represent a group of structurally diverse molecules, ranging from persistent pollutants to microbiome metabolites. Sustained AHR activation by cigarette smoke exposure may even increase TED pathology and could explain why cigarette smoking is the biggest modifiable risk factor for the disease. 59 In contrast to sustained and chronic activation of AHR by pollutants like dioxin and cigarette smoke, FICZ and microbiome metabolites such as indole-3-aldehyde (I3A) only transiently activate the AHR resulting in homeostatic or even beneficial effects. 60 A recent study identified an altered gut microbiome in both Graves’ disease and TED. 61 The gut microbiome is also different in mice presenting symptoms that mimick TED, further supporting the potential role of the microbiome in TED. 62,63 Future studies are needed to evaluate if changes in the microbiome alter endogenous AHR signaling, potentially exacerbating disease phenotype.
AHR activation by FICZ significantly upregulates the expression of SOD3, an enzyme that neutralizes superoxide radicals. SOD3, unlike other SODs, is localized to the extracellular space and protects the extracellular matrix from oxidative damage. SOD3 overexpression prevents bleomycin-induced tissue remodeling, pulmonary hypertension, and expression of TGFβ in a mouse model of lung fibrosis. 64 Further, SOD3 expression blocked cell migration by controlling cellular adhesion molecules and inflammatory cytokine production in animal models of peritonitis and ischemia. 47
IGF1R activation-induced expression of tubulin genes and CRABP2. To our knowledge, this is the first report showing that IGF1R activation can induce CRABP2 expression. CRABP2 can potentially regulate the expression of many genes by binding, sequestering, or transporting retinoic acid to retinoic acid receptors. 65 The role of CRABP2 in TED OFs is still unclear and requires further study. In another potential mechanism, CRABP2 activates proliferation through β-catenin signaling in dermal papilla cells. 66
Our study has revealed several promising findings with therapeutic potential for TED treatment. We have demonstrated that AHR signaling can effectively counteract IGF1R-mediated pathological processes in TED OFs through regulation of the GSK3β pathway. The activation of GSK3β by AHR ligands may have broader therapeutic implications beyond IGF1R inhibition, as GSK3β also regulates other disease-relevant pathways including TGFβ signaling 31 and adipogenesis. 67 These findings are particularly significant for patients who may not respond to or tolerate conventional therapies, including teprotumumab. The identification of AHR as a potential therapeutic target opens new avenues for drug development, especially for patients with limited treatment options. Future studies exploring the therapeutic potential of AHR ligands, delivery routes, and selective ligands that produce transient activation could lead to novel treatment strategies for TED that simultaneously target multiple pathological pathways.
Footnotes
Acknowledgments
The authors thank John Ashton and the University of Rochester’s Genomics Research Center for creating the RNA-seq data. The authors are grateful to tissue donors who have generously shared their tissue with us.
Authors’ Contributions
E.R.: Data curation (lead), methodology, analysis, review, and editing (equal). F.H.: Data curation, methodology, analysis, and writing—review and editing (equal). C.P.P.: Data curation and writing—review and editing (equal). S.E.F.: Conceptualization, methodology, analysis, and writing—review and editing (equal). C.F.W.: Conceptualization (lead); methodology, data curation, analysis, writing—original draft (lead), and writing—review and editing (equal).
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This research was supported by the National Institutes of Health Grant (EY031398) is to C.F.W. The Unrestricted Grant from the Research to Prevent Blindness is to the University of Rochester Medical Center Department of Ophthalmology.
Supplementary Material
Supplementary Data
Supplementary Figure S1