
Abstract
Background:
Thyroid-associated ophthalmopathy (TAO, aka thyroid eye disease [TED], Graves’ orbitopathy) remains poorly understood and inadequately treated since its initial description. It is disfiguring, can threaten vision, and represents an autoimmune process closely associated with thyroid disease. Unambiguous connections linking TAO to the glandular maladies of Graves’ disease (GD) remain incompletely clarified. Detecting the thyrotropin receptor (TSHR) in periocular tissues suggests that this cell-surface protein represents a shared autoantigen with the thyroid gland, but we now know that its expression is ubiquitous. Most patients with TAO have relatively high circulating levels of activating anti-TSHR autoantibodies. Emerging more recently is the importance of insulin-like growth factor I receptor (IGF-IR) in the pathogenesis of TAO. The TSHR/IGF-IR signaling complex apparently drives circulating fibrocytes and the unique phenotypes of fibroblasts inhabiting the TAO orbit (GD-OF).
Methods:
The PubMed database was scanned for articles dating back to the earliest time periods covered. Keywords used for primary searches included thyroid-associated ophthalmopathy, Graves’ orbitopathy, TED, orbit, TSH receptor, IGF-I receptor, and autoimmune thyroid disease. Secondary searches used numerous other search terms.
Results:
GD-OF have been characterized extensively as being particularly responsive to the immunological factors and key effectors in TAO pathogenesis. Both TSHR and IGF-IR are overexpressed by GD-OF and CD34+ fibrocytes and form a signaling complex. They are activated through this TSHR/IGF-IR complex to produce large amounts of hyaluronan and express multiple cytokines. This complex mediates cellular responses to pathogenic IgGs in TAO. CD34+ fibrocytes and CD34+ OF also express relatively high levels of multiple thyroid autoantigens. Identifying IGF-IR as a key component of a receptor complex and its intertwining signaling activities with those of TSHR has led to a targeted medical therapy for TAO. This therapy involves the selective systemic inhibition of IGF-IR.
Conclusions:
Much has been learned over the preceding decades about the pathogenesis of TAO. Among these is the identification of IGF-IR as a pivotal component underpinning the disease. This has led directly to development of an effective targeted therapy. Important gaps in our understanding persist, and current therapies have limitations. Thus, despite these advancements, considerably more remains to be achieved.
Introduction
Thyroid-associated ophthalmopathy (TAO, aka thyroid eye disease) represents a presumed autoimmune disease in which connective tissues inhabiting the orbit and upper face become inflamed and undergo remodeling that can result in ocular dysfunction and reduced quality of life. 1 TAO is disfiguring and can lead to vision loss. It is closely associated with autoimmune diseases of the thyroid gland, most frequently Graves’ disease (GD) and less commonly with Hashimoto’s thyroiditis. 2 The mechanistic links between TAO and thyroid gland autoimmunity remain in large part speculative and are the topics of substantial debate. Controversies span from the contested interpretation of experimental results to disagreement about the most proper nosology and nomenclature that best describe TAO. In this study, I attempt to present widely divergent opinions about the pathogenesis of TAO, acknowledging both the strengths and weaknesses of multiple perspectives. In particular, the article focuses on the putative roles played by thyrotropin receptor (TSHR) and insulin-like growth factor I receptor (IGF-IR) and the cell types that express these proteins relevant to TAO (Fig. 1). The disease process involves numerous molecular effectors, including cytokines, growth factors, and lipid mediators. As should become evident, major gaps in our understanding of this complex disease process remain.
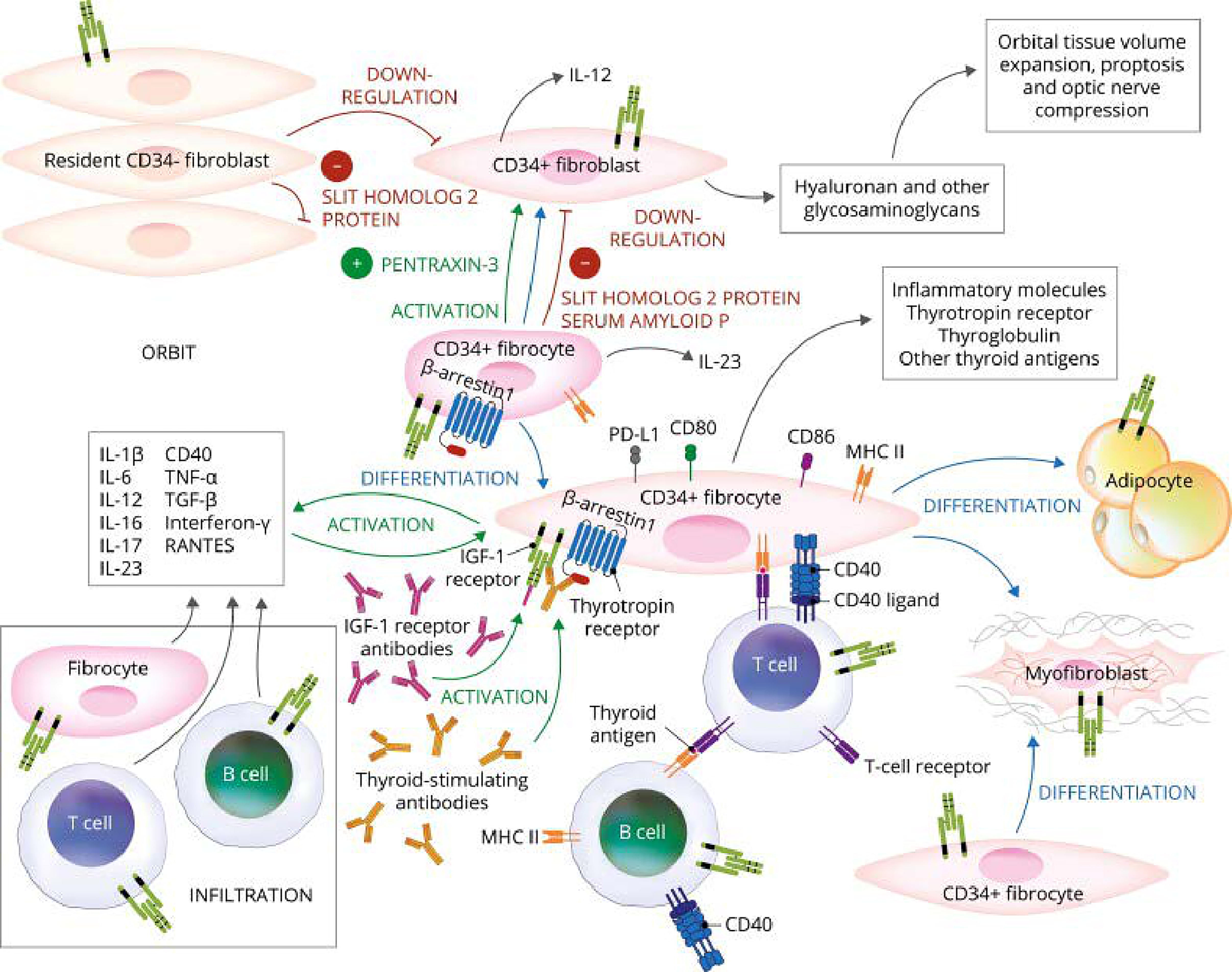
Theoretical model of thyroid-associated ophthalmopathy (TAO) pathogenesis and the central involvement of TSH and IGF-I receptors. Orbital fibroblasts exhibiting robust responses to inflammatory mediators appear to represent the central effector cells. CD34+ orbital fibroblasts derived from CD34+CXCR4+Collagen 1+ fibrocytes, monocyte-lineage progenitors, traffic from bone marrow to the TED orbit. Fibrocytes express several thyroid-specific proteins, including thyrotropin receptor (TSHR), thyroglobulin, thyroperoxidase, and sodium–iodide symporter, and Class II major histocompatibility complex (MHC). Fibrocytes and orbital fibroblasts undergo differentiation into myofibroblasts and adipocytes. Slit2 expressed and released by CD34− fibroblasts downregulate expression of many genes expressed by fibrocytes and CD34+ fibroblasts. Interleukins 1β, 6, 8, 10, 12, 16, 23, tumor necrosis factor α, and regulated on activation, normal T expressed and secreted (RANTES), CXCL-12 and CD40-CD154. CD34+ and CD34− orbital fibroblasts cell surface display IGF-I receptor and express three mammalian hyaluronan synthase isoenzymes and UDP glucose dehydrogenase and synthesize hyaluronan. This glycosaminoglycan underlies, in part, orbital tissue expansion in TED. Hyaluronan synthesis localizes primarily to CD34− orbital fibroblasts. Reprinted from Smith TJ. J Clin Endocrinol Metab 2022 107:S13-S26 (with permission).
A Search for Antigens Involved in TAO
Identification of the initial pathogenic instigators of TAO has long been sought. Because of its clinical presentation, many of the early studies focused on orbital fibroblasts from patients with TAO (GD-OF), serum autoantibodies detected in patients with GD, and various lymphocyte populations. Exposure to multiple thyroid antigens can activate both T cell (Tc) and B cell (Bc) responses in states of thyroid autoimmunity. These autoantigens include TSHR, 3 but whether Tc and Bc activated by thyroid antigens play roles in TAO analogous to those occurring in the thyroid manifesting GD remain uncertain. Several candidate orbital autoantigens relevant to TAO have been proposed previously. These include a 64 kDa extraocular muscle (EOM) membrane protein. 4,5 Examination of Tc responses suggested that this 64 kDa protein is unlikely to be relevant to TAO pathogenesis. 6 A 23 kDa soluble fibroblast protein is recognized by antibodies in sera from patients with GD. 7 The protein can be detected in fibroblasts from multiple anatomical regions, regardless of whether the donor is healthy or manifests TAO. Alternatively, antigen “sharing” between the thyroid gland and orbit has long been postulated and remains plausible. The concept that thyroglobulin (Tg) might represent such a shared autoantigen in TAO was introduced by Kriss and colleagues. 8,9 They postulated that the source of orbital Tg could be traced to the thyroid, but this concept remained unproven. The source of orbital Tg appears to have been solved when CD34+ fibrocytes, monocyte progenitors, were identified in diseased orbital tissue. 10 –12 These cells and their CD34+ GD-OF derivatives express functional Tg and TSHR, 13 as well as thyroperoxidase and sodium–iodide symporter. 14 Expression of these thyroid antigens in fibrocytes (high level) and GD-OF (low level) is driven by the autoimmune regulator protein. 14 Disparity of the expression levels is the consequence of Slit2. 15,16 Whether these autoantigens, besides TSHR, play any part in the development of TAO is uncertain. Finding that tissue-infiltrating cells such as fibrocytes express these proteins at meaningful levels raises potentially important issues regarding how unexpectedly widespread they are and now raise questions about their biological consequences beyond the thyroid. Furthermore, we currently have no information regarding whether particular antigen-specific Tc against thyroid antigens are involved in orbital tissue remodeling in TAO.
The role of intraorbital Tc in TAO pathogenesis may involve direct cross talk with GD-OF through the molecular conduit of CD40/CD154, 17,18 resulting in the induction of multiple inflammatory cascades and robust hyaluronan (HA) synthesis. 19,20 It remains uncertain whether autoreactive, antigen-specific Tc are among those infiltrating the orbit and whether Tc and Bc infiltration of the orbit varies as a function of TAO duration, clinical behavior, or treatment. Six interleukin (IL)-2-expanded Tc lines collected from TAO tissues were dominated by the CD8+CD45RO+ phenotype and, when activated, secreted IL-4, interferon (IFN)-γ, and IL-10. All lines proliferated in response to autologous retrobulbar fibroblasts in the absence of cytotoxic activities. 21 In vitro-expanded Tc infiltrating EOM were predominantly CD4+CD45RO+ producing multiple cytokines of which IL-4 was invariably detected. 22 Importantly, thyroid tissue extract elicited a proliferative response in these EOM infiltrating Tc in vitro. In some studies, orbital Tc have a Th1 cytolytic phenotype and secrete IL-2, IFN-γ, and tumor necrosis factor α. 23,24 Other investigators found IFN-γ expression to be absent but detected IL-4 mRNA in a majority of patients. 25 Comparing orbital Tc from short versus long duration TAO revealed that earlier (shorter duration) TAO was associated with Th1 cells, while longer duration disease (>2 years) was associated with Th2 cytokine production. 26 Thus, the divergence among TAO cases might correctly be attributed to disease duration rather than other factors. Further studies will be required as follows: (1) to identify the precise roles of TSHR and other thyroid proteins and antigen-specific Tc in TAO and (2) to clarify whether disease duration imposes bias of Tc polarization and expansion and whether this phenomenon might be harnessed as a clinically useful assessment in TAO.
TSHR and Its Role Specifically in TAO
TSHR is a ubiquitously expressed G protein coupled receptor (GPCR) (Fig. 2), the activity of which is the major determinate of thyroid gland growth and hormone biogenesis. 27 –30 Many of the initial mechanistic studies of how it functions as an activator of adenylate cyclase were conducted in culture models of epididymal fat pads, cerebral cortex, and liver tissues exposed to purified bovine TSH. 31 Its role in the pathogenesis of GD was recognized more than 50 years ago, 32 when Adams and Purves described long-acting thyroid stimulator in sera from hyperthyroid patients with GD. 33 Thus, at the center of thyroidal GD development is the loss of immune tolerance to TSHR 32 and the generation of activating anti-TSHR autoantibodies (thyroid-stimulating immunoglobulins, TSIs). 34 These activating IgGs are admixed with those blocking TSHR 35 or binding to the hinge region but failing to elicit canonical responses and, instead, activating reactive oxygen species. 36 Autoantibodies generated against TSHR are heterogeneous with diverse binding characteristics and biological effects on thyroid function. 37 The central function of TSHR, both in health and disease, is established, and thus, therapeutic development for treating thyroid dysfunction in GD by interrupting its inappropriate activity is well-conceived. It remains uncertain why patients with high levels of circulating TSH, most frequently in primary hypothyroidism, fail to manifest TAO. Potential explanations for this divergence include the responses peculiar to polypeptide molecules versus those of antibody agonists involving G-protein coupled functions and activation of noncanonical GPCR signaling. Another concerns the differences in cell-signaling molecules (such as β-arrestins, β-Arr) found in different tissues and cell types (Fig. 2). 38
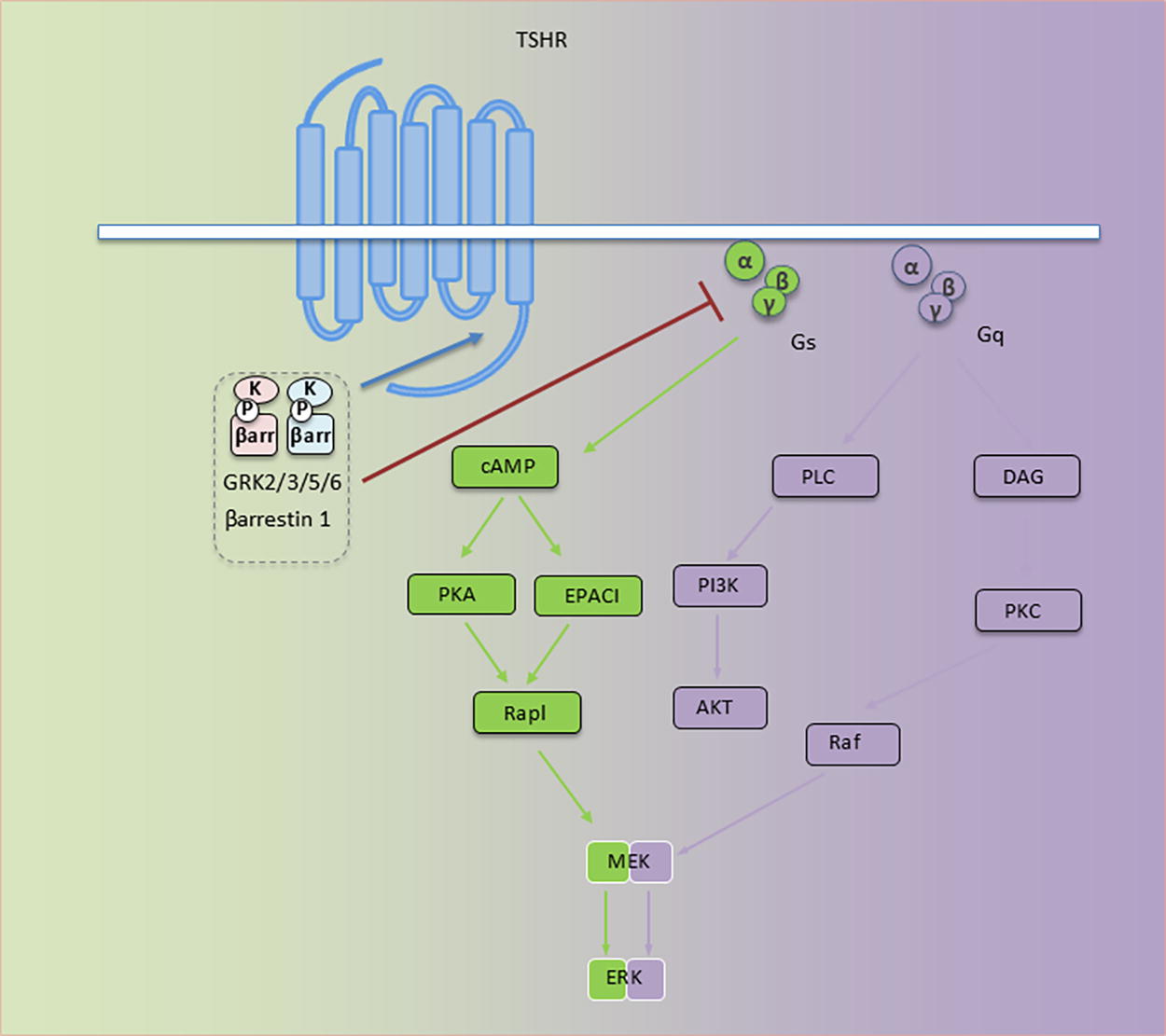
TSH receptor (TSHR) signaling. Major signaling pathways utilized by TSHR. Under TSHR inactive TSHR states, Gα (α) is bound to guanosine diphosphate (GDP) and Gβγ (βγ). This forms an inactive Gs or Gq containing trimer, αβγ. Activated TSHR results in GDP/GTP exchange on the α subunit and dissociation into α and βγ subunits. α and βγ subunits then interact with second effector proteins, promoting downstream pathway activation; TSHR/Gs activates adenylyl cyclase (AC) and an increased cAMP level. Generated cAMP then regulates several signaling pathways, among them ion channels, PKA and EPAC. TSHR/Gq activates phospholipase C (PLC)/phosphoinositide 3-kinase (PI3K) cascade, thus promoting cell proliferation and survival. TSHR/Gq enhances diacyl glycerol (DAG) generation and protein kinase C (PKC) activation. Gs and Gq activate Raf/MEK/ERK (mitogen-activated protein kinase) pathways. βγ subunits initiate receptor-distinct signaling, including G protein-coupled receptor kinase (GRK) recruitment. Phosphorylation of serine/threonine residues by GRKs occurs within the TSHR C-terminus. These create β-arrestin (β-arr) binding sites. β-arr recruitment prevents further G protein activation (i.e., desensitization). All signaling pathways downstream from TSHR actuate biological effects through transcription factor activation. Reprinted from Girnita et al. J Clin Endocrinol Metab 2022 107:S1-S12 (with permission).
By logical extension, a similar importance of TSHR and the autoantibodies generated against it has been ascribed to TAO. Supporting this concept are several findings that either directly or indirectly make this a compelling link as follows: (1) the notable detection of both TSHR-encoding mRNA and receptor protein in orbital tissues. 39 –41 Levels of expression were detected in orbital fat from patients with active TAO being higher than those with stable disease. 42 TSHR mRNA and protein were subsequently detected in cultured orbital fibroblasts from patients with GD (hereafter referred to as GD-OF). 43 These expression levels were considerably lower than those found in thyroid epithelium. 44 Uncertainty surrounds whether these differences carry important biological consequences. Contravening the concept that TSHR and autoantibodies generated against it comprise a monolithic autoimmune response pathway underlying TAO is the ubiquitous expression of this receptor. Many of the tissues/cell types inhabit anatomical regions not overtly manifesting GD. 40,45 –51 (2) GD-OF respond in vitro to TSH, admixtures of GD-IgGs, and the well-characterized TSHR-activating mAb, M22. 10,52 These responses suggest a potential functional role for TSHR in the orbit. 44 TSH can enhance GD-OF proliferation 53 and HA synthesis 54 and increase cytokine expression/production, including IL-1β, IL-1 receptor antagonist, IL-6, IL-12, IL-16, and IL-23; 16,55,56 (3) Higher TSI levels are found in patients with active/severe TAO compared with those with GD either without clinically apparent TAO or with mild/inactive orbital involvement. 57,58 (4) Approximately 90% of patients developing TAO have already developed clinically recognizable autoimmune-driven thyroid dysfunction or will do so in the future. Thus, a strong case has been made for TSIs driving TAO pathology through TSHR activation in a manner analogous to their role in the thyroid gland over- (or under-) activity characteristic of GD. The positive correlation between TAO and anti-TSHR antibody levels has been generated when certain autoantibody assays were used, but not with others. 59 Furthermore, this relationship has not been a consistent finding in all reports. 60 Correlations between TSHR-simulating autoantibody levels and the clinical behavior of TAO were found superior to those of binding inhibitory immunoglobulins (TBII). (5) Immunization of mice with TSHR results in a mouse phenotype resembling some but not all features of GD and TAO. 61 –64 Thus, a substantial body of evidence supports the involvement of TSHR in the pathogenesis of TAO.
TSHR as a Plausible Therapeutic Target for TAO
Given the established role for TSHR in the pathogenesis of GD, multiple programs have actively pursued development of therapeutics targeting that receptor. These include efforts toward identifying or creating small molecules 65 and monoclonal antibodies, 66 as well as efforts to restore immune tolerance to TSHR, 67 a putative defect in thymic expression of the receptor. 68 Recent preclinical studies have revealed inhibition by TSHR ligands of cancer cell proliferation. 69 Blocking antigen-Tc interactions based on a personalized genetic background is an attractive approach to managing autoimmune thyroid disease. 70,71 More evidence will be necessary before one can fully evaluate the therapeutic potential of these strategies directed at TSHR.
IGF-IR, Its Ubiquitous Involvement in Cellular Functions and Insinuation into TAO
Since its molecular cloning, development of sensitive and specific assays for detecting anti-TSHR autoantibodies has been successfully undertaken. While these autoantibodies have been detected in sera from a vast majority of patients with TAO, those specimens from a few affected individuals have proven negative for detectable anti-TSHR autoantibodies. One early study demonstrated that IgGs from a TBII-negative patient could nonetheless stimulate thyroid activity. 72 The early nature of this report (1991) suggests that potential limitations of the TBII assay used could account for these antibody-negative findings. In any event, the rare patient with TAO in whom anti-TSHR antibodies are undetectable 73 –75 has invited consideration of other autoimmune targeted pathways being involved in the orbital disease. Differences in assay sensitivity/technique may ultimately provide the basis for these infrequent discrepant findings. Thus, some investigators have questioned whether antibodies generated in GD might target other antigens and perhaps whether they are capable of activating tissues. Among the more recent candidate autoantigens relevant to TAO is IGF-IR.
Cell surface displayed tyrosine kinase receptors (RTKs), exemplified by IGF-IR, are expressed widely in virtually every mammalian cell type and tissue. 76 Their signaling patterns are extremely complex and involve both canonical and noncanonical mechanisms 38 (Fig. 3). Initially known as somatomedin C receptors, 77 they frequently comprise a physical protein complex with the insulin receptor. The two receptors share substantial structural similarity and postreceptor signaling pathway usage but are known to possess divergent mitogenic and metabolic regulatory profiles. 78,79 The patterns of IGF-IR dimerization determine the nature of signal activation emanating from the receptor. Detailed reviews of IGF-IR characteristics have appeared recently, including their potential involvement in autoimmune diseases and cancer. 76,80
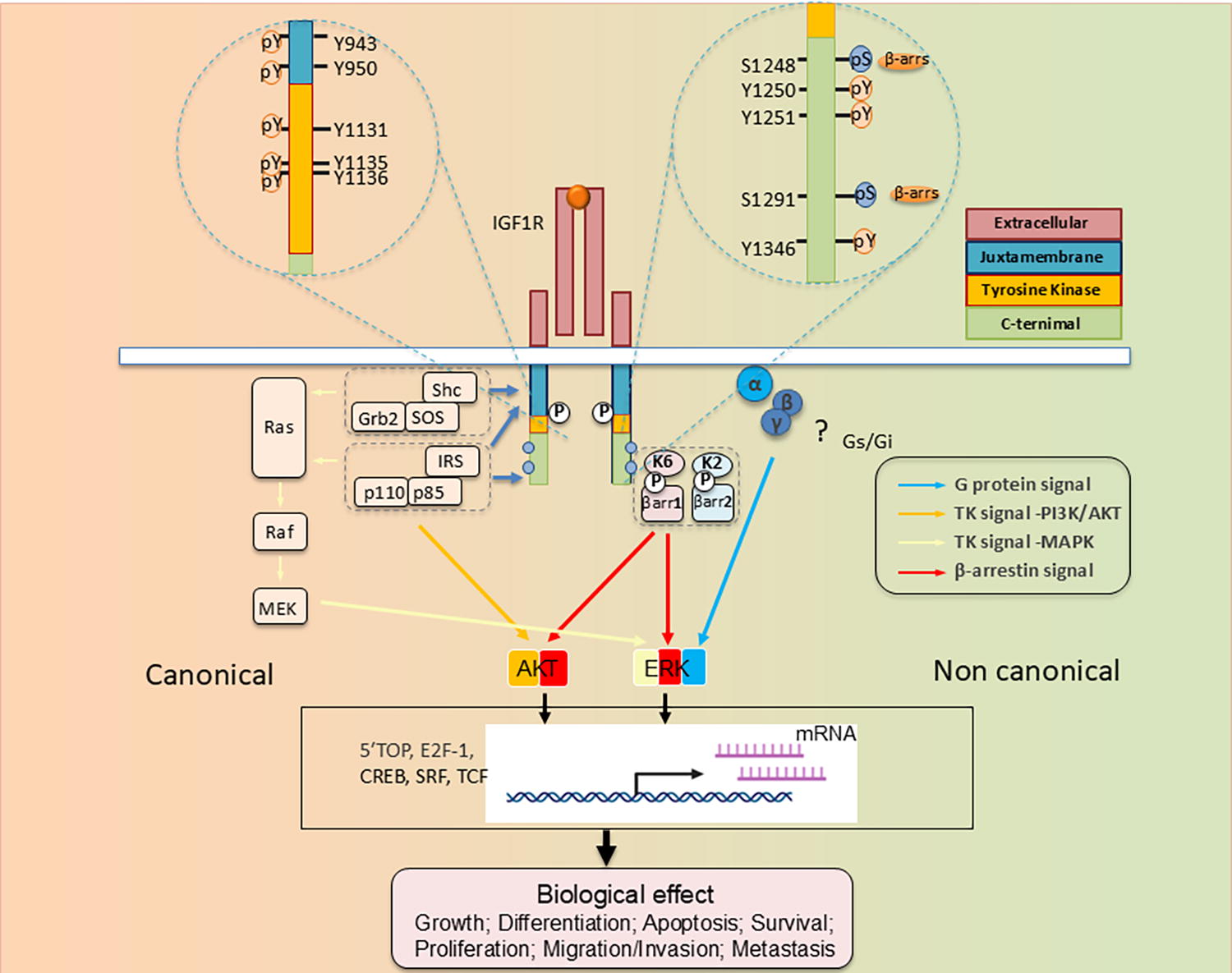
Structure–function relationship of IGF-I receptor (IGF-IR) informing tyrosine kinase receptors (RTK)/G protein coupled receptor (GPCR) duality. IGF-IR is annotated with numbered amino acid residues. Thus far identified residues/sites post-translationally modified (PTM) as determinant substrates/adaptor protein binding within the receptor β-subunit. These determine both canonical RTK signaling (left) and noncanonical signaling resembling that of GPCRs. Kinase-dependent IGF-IR signaling pathways: IGF-1 and IGF-II binding to the receptor elicits intrinsic tyrosine kinase activity and auto-phosphorylation. Activated IGF-IR then recruits and phosphorylates substrates, including insulin receptor substrate (IRS) and SHC-transforming protein (Shc). pY 950 within the NPXY juxta-membrane motif represents an essential component enabling IRS-1, IRS-2, and Shc recruitment. Tyrosine phosphorylation of IRS and Shc proteins leads to binding of Grb2 and PI 3-kinase (p110/p85). This binding induces multiple downstream signaling activation events, primarily involving Ras/Raf/MAPK and PI3K/Akt. The transcription factors involved in IGF biological effects are consequently activated. Besides intrinsic IGF-IR kinase-dependent signaling, agonist-induced receptor stimulation can result in noncanonical GPCR signaling through heterotrimeric G-proteins (α, β, γ) (“?” denotes those pathways/events not yet fully understood), followed by rapid receptor phosphorylation through actions of G-protein-coupled receptor kinases (GRKs, K2, K6) at the C-terminus containing serine residues. Serine-phosphorylated receptors present high affinity binding sites for the adaptor proteins, β-arrestin 1/2 (β-arr1/2). β-arr assumes an active conformation following IGF-IR binding to MAPK pathway components, resulting in “second wave” IGF-IR kinase-dependent MAPK/ERK signaling. “P” denotes key phosphorylation sites; Akt, protein kinase B; ERK, extracellular-signal-regulated kinase; IGF-IR, insulin-like growth factor-I receptor; IRS, insulin receptor substrate; MAPK, mitogen-activated protein kinase; MEK, mitogen-activated protein kinase/Erk kinase; Shc, Src homology and collagen domain protein. Reprinted from Girnita et al. J Clin Endocrinol Metab 2022 107:S1-S12 (with permission).
Whether IGF-IR plays an important role in TAO pathogenesis remains widely disputed as does the apparent generation of anti-IGF-IR autoantibodies in patients with GD and TAO. The evidence supporting IGF-IR involvement, dating back several decades, includes the findings of Kendall-Taylor and colleagues demonstrating that GD-IgGs recognize binding determinants on GD-OF plasma membranes. 81 Their evidence, suggesting that autoantibodies targeting IGF-IR are generated in patients with TAO, went untouched for several years before further investigations into these binding sites were reported. Studies by Pritchard et al. established that IGF-IR and not IGF-I binding proteins (IGFBPs) accounts for the saturable GD-IgG and IGF-I binding to GD-OF. 82 Those studies and others that followed suggested that anti-IGF-IR autoantibodies could activate the synthesis and release of chemoattractants from GD-OF, including IL-16 and regulated on activation, normal T expressed and secreted (RANTES). 82,83 Among the strongest evidence supporting the generation of these anti-IGF-IR antibodies is their apparent detection in sera from patients with rheumatoid arthritis (RA). 84 Pritchard et al. reported that IgGs derived from patients with RA could also induce cytokine production in both GD-OF and RA synovial fibroblasts. 84 Smith and Hoa 85 demonstrated that GD-IgG and IGF-I could induce HA synthesis and release from GD-OF. In contrast, no such responses to rhTSH could be detected. While disease-derived cells responded to IGF-I and GD-IgG, their healthy donor-derived fibroblast counterparts failed to do so. Thus, those studies demonstrated a fundamental difference between disease-derived cells and those from healthy tissues. Specifically, cell-surface IGF-IR density is substantially greater on GD-OF than that found on fibroblasts derived from healthy tissue. 44
Subsequent reports have yielded a variety of findings without consistent evidence for or against meaningful involvement of IGF-IR or the autoantibodies targeting that receptor in TAO. Marino et al. detected anti-IGF-IR autoantibodies (function undetermined) more frequently in patients with GD (25% vs. 0% in healthy controls), regardless of whether they manifested TAO. 86 A subsequent report from that group suggested that these antibodies were more frequently detected in patients with GD without TAO than in those with the orbital disease. 87 In contrast, Varewijck et al. reported GD-IgG that stimulate IGF-IR activity in a subset of patients with TAO, 88 while Krieger et al. failed to detect IGF-IR stimulating autoantibodies. 89 Those investigators have long contended that any biological effects of GD-IgGs result from direct TSHR activation. 90 They used supraphysiological concentrations of IGF-I (100 ng/mL) as the comparator for their studies assessing GD-IgG stimulation of phosphorylated IGF-IR as the only cell response readout. They concluded that any effects of GD-IgGs targeting IGF-IR are “modest.” Absent from their studies were comparisons of responses to GD-IgG with those of physiological concentrations of IGF-I. In addition, detailed time-course studies (they used a single timepoint) were absent, thus leaving potentially time-sensitive responses undetected. Furthermore, they failed to evaluate IGF-IR tyrosine kinase-independent activity such as that initiating β-Arr-biased signaling. These variable findings in studies examining potential responses to anti-IGF-IR autoantibodies could have also resulted from differences in experimental conditions. Variables include serum IGF-I and IGFBP concentrations in preincubation media, culture monolayer confluence, and the GD-IgG treatment durations. It should be stressed that in many of these studies, intrinsic tyrosine kinase activation was the single response readout, ignoring potential responses mediated through alternative pathways. In sum, the evidence for and against the generation of activating anti-IGF-IR autoantibodies in patients with GD and TAO remains inconclusive. Further studies, ideally using standardized conditions and sensitive assays, should settle unambiguously this persisting controversy.
In addition to in vitro studies involving human cells, evidence that anti-IGF-IR autoantibodies might be generated as a consequence of immunization to TSHR in rodents has been reported. In a series of studies where female BALB/c mice were immunized with hTSHR A and IGF-IR α subunits delivered as muscle electroporated plasmids, autoantibodies directed against both TSHR and IGF-1R were detected in those mice receiving a single immunization with the hTSHR-encoded plasmid. In contrast, anti-hTSHR A autoantibodies were absent in animals receiving either IGF-IRα or control plasmids, and these animals failed to develop a TAO-like phenotype. 62,63 It should be noted that many of those mice immunized with hTSHR A and generating detectable anti-IGF-IRα autoantibodies developed disease-resembling phenotypic attributes. Thus, generation of autoantibodies against IGF-IRα following immunization with hTSHR A alone in mice suggests cross-antigenicity shared between the two receptors, either as the consequence of epitope spreading or some other, yet undefined immune response mechanism. A very recent study by Wu et al. found that co-immunization with TSHR and IGF-IR enhanced the development of TAO compared with those immunized with TSHR alone. 91
Molecular Collaboration Between TSHR and IGF-IR
TSHR has long been identified as interacting with other GPCRs, including the A1 adenosine receptor. 92 Comingling of pathways downstream of GPCRs and RTKs was recognized by Lefkowitz and colleagues nearly 30 years ago. 93 Identifying this interplay between structurally and functionally dissimilar receptors has added to our understanding of the complexities associated with cell regulation. 94 –97 IGF-IR, a prototypic RTK, can collaborate with GPCRs (Fig. 4). 98 Thus, the interactions between TSHR and IGF-IR should not have been surprising. The seminal observations by Ingbar and colleagues demonstrated that IGF-I and insulin could both enhance the biological responses of TSH in thyroid epithelial cells in vitro. 99,100 Using rat thyroid epithelial FRTL-5 cells, they reported that IGF-I could upregulate the mitogenic activity of either TSH or GD-IgG. TSHR activation of GD-OF with bovine TSH is enhanced by cotreatment with IGF-I, 101 suggesting that the relationship between the two pathways is generalized accross multiple cell types. Aggressive thyroid cancers express IGF-II, 102 postulated to represent a factor involved in cellular dedifferentiation. GD-OF overexpress IGF-IR when compared with orbital fibroblasts from donors without TAO. 82 Other reports disclosed that GD-OF secrete higher levels of IGF-I 103,104 and express elevated levels of somatostatin receptor 2104 when compared with healthy donor-derived fibroblasts. Furthermore, octreotide was found to attenuate IGF-I production and release from GD-OF. Thus, multiple components of the IGF-I pathway are upregulated in TAO and that GD-OF exhibit fundamental differences when compared with those orbital fibroblasts from healthy individuals.
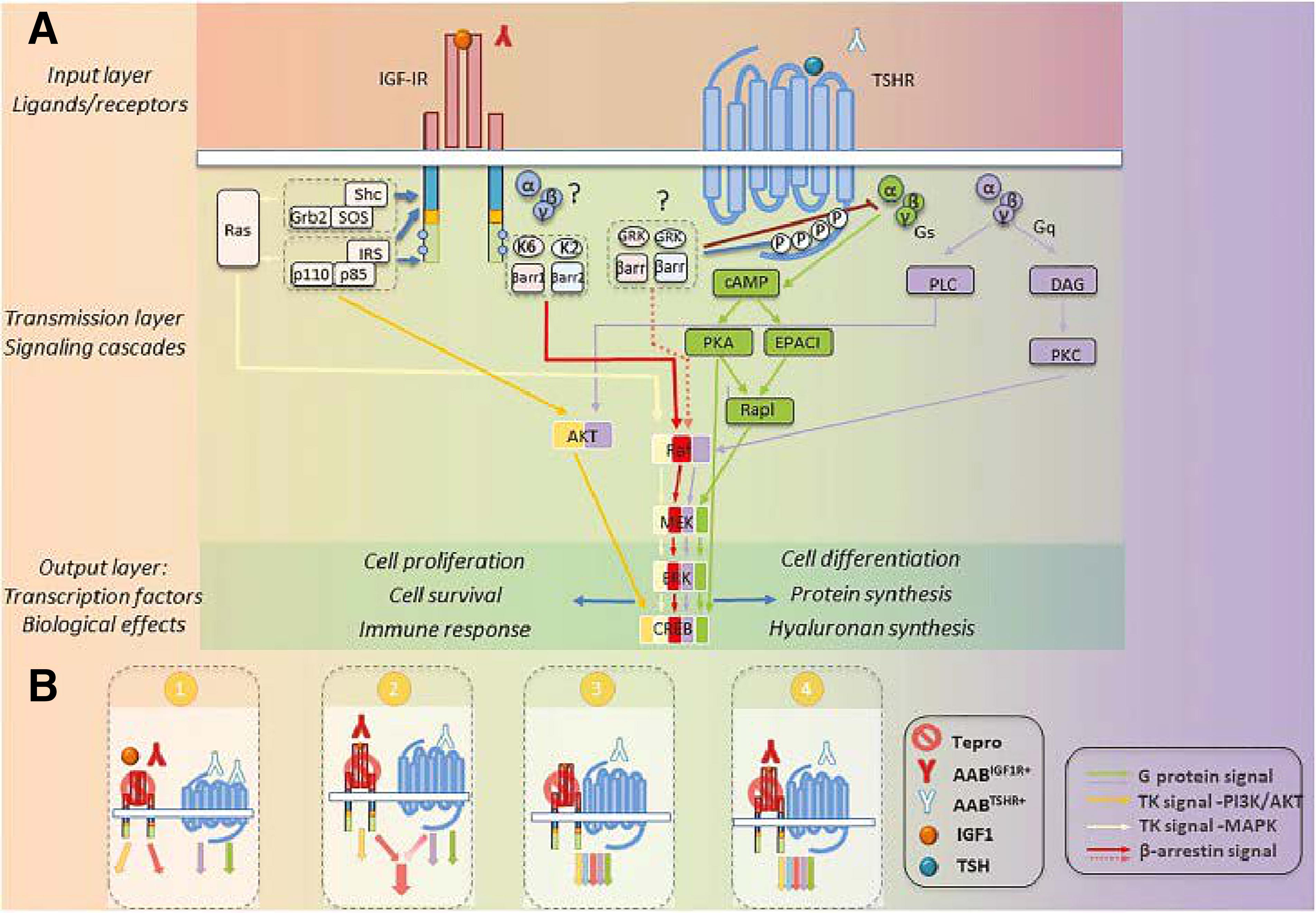
Those observations of Ingbar and colleagues concerning the enhancement of responses to TSH by IGF-I and insulin lay fallow for two decades. Then Tsui et al. demonstrated that TSHR and IGF-IR colocalize in cultured GD-OF, primary human thyroid follicular cells, and in situ in orbital connective tissues, when visualized by confocal microscopy. 44 Those studies revealed that the two receptor proteins coimmunoprecipitated, since they could be pulled out of solution with either anti-TSHR or anti-IGF-IR mAbs. This result suggested strongly that they are physically linked. Further studies knocking down IGF-IR expression with a dominant negative receptor or inhibiting its activity with 1H7, 44 a mAb targeting IGF-IR, 105 could block Erk activation in primary thyroid epithelial cells by human recombinant TSH, GD-IgG, or IGF-I. 44 Thus, the authors suggested that therapeutically targeting IGF-IR might represent a “two-shots on goal” strategy for interrupting pathogenic signaling initiated at either TSHR or IGF-IR. They envisioned that IGF-IR attenuation might represent a treatment for TAO. 82 Additional studies disclosed that other autoimmune diseases, such as RA, might share anti-IGF-IR autoantibody generation. 106
Following the report of Tsui et al. 44 that IGF-IR collaborates with TSHR, other groups followed suit. One article demonstrated that M22 treatment of GD-OF resulted in biphasic responses using HA secretion as the readout. 107 In that study, an IGF-IR antagonist blocked the higher but not the lower “potency” response. M22 failed to elicit intrinsic IGF-IR tyrosine phosphorylation, as would be anticipated a priori. These authors concluded that cross talk between the two receptors depends on TSHR activation and subsequently tested 57 samples of GD-IgG, reporting that none resulted in detectable IGF-IR tyrosine kinase activation. 89 Another laboratory group found that GD-IgG inhibited the activation of IGF1R tyrosine phosphorylation by IGF-I. 108 Yet another interpreted their experimental results as demonstrating that sera from a subset of patients with TAO could stimulate IGF-IR tyrosine phosphorylation, activity that they could abolish by depleting GD-IgG from the serum samples. 88
Thus, controversy surrounds whether TSHR, the central autoantigen driving the hyperthyroidism in GD, represents the monolithic cell surface receptor integral to TAO development. IGF-IR represents a more recently identified and apparently important piece of the TAO puzzle.
Teprotumumab Emergence as a Highly Effective Therapy for TAO
Therapeutic targeting of IGF-IR in cancer represented an active research focus two decades ago. 109 Despite substantial plausibility, several mAbs and small molecules developed by multiple cancer-targeting therapeutic programs failed clinical efficacy trials, rendering them available for repurposing for other diseases. Studies performed entirely in vitro using GD-OF and related cell types, including CD34+ fibrocytes, Tc, and Bc, ushered in the development of teprotumumab as specific therapy for TAO. 110 Despite substantial evidence, whether IGF-IR might, in addition to TSHR, represent a sensible therapeutic target for TAO remains controversial. Among that hypothesis-testing evidence to date, two pivotal, randomized, placebo-controlled trials of patients with active (clinical activity scores ≥4), moderate-to-severe TAO were treated with 8 infusions over a 24-week period. 111,112 The primary outcome was responder rate of proptosis reduction ≥2 mm from baseline in the more severely affected eye (p < 0.0001 vs. placebo in both studies). Secondary outcomes (overall CAS + proptosis responder rate, subjective diplopia improvement, and quality of life [functional scale]) were also highly significant. Subsequent trials, including a longer disease duration and retreatment extension (OPTIC X) of the phase 3 trial, 113 and a long-disease duration and stable TAO trial supported effectiveness of teprotumumab. 114,115 Since its approval by the U.S. Food and Drug Administration in January 2020, over 20,000 patients have been treated with teprotumumab. Accumulating evidence indicates that the drug is effective in longer duration, lower CAS activity TAO, and with retreatment for incompletely treated/recrudescent disease. 113 –116 Thus, the evolving experience with teprotumumab, in the context of both formal clinical trials and in real-life practice, strongly suggests a continued role for IGD-IR inhibition in our treatment paradigms for TAO.
Adverse Events Associated with Teprotumumab Therapy for TAO
While targeting IGF-IR with teprotumumab, an inhibitory monoclonal antibody, has already proven to be effective, the side effect profile emerging from pivotal phase 2 and phase 3 clinical trials had revealed adverse events (AEs) concerning hearing, hyperglycemia, menstrual irregularities, and muscle cramps among others, largely judged as mild to moderate. 111,112 Subsequent real-world reports have also revealed many of these same AEs. These are in keeping with the anatomically diverse side effects often encountered with therapeutic targeting of ubiquitously expressed molecules such as IGF-IR. Since its entrance into clinical practice, several reports of AEs putatively associated with teprotumumab treatment in real-world settings have appeared. A study cohort of 131 patients treated with teprotumumab across 6 centers was evaluated for AEs. 117 Mean follow-up time was 70.2 ± 38.5 weeks after the first infusion. Of those included, 81.7% of patients experienced at least one AE (median of 4 per patient), and 12.2% (16/131) discontinued treatment because of one or more AEs, including hearing loss (n = 4), inflammatory bowel flare (n = 2), hyperglycemia (n = 1), muscle spasms (n = 1), and multiple AEs (n = 8). The limitations of this study have been opined on previously. 118 As with virtually all novel therapeutic approaches, teprotumumab has attracted concerns, most frequently arising from its apparent association with hyperglycemia 119 and hearing abnormalities. 120,121 Rates of AE occurrence recognized from the clinical trials have differed from those in some postmarketing reports, none of which was placebo controlled or used standardized adjudication.
The intimate relationship between IGF-I and insulin pathways, including their respective receptors and postreceptor signaling, 122,123 underlies why systemic therapeutic inhibition of IGF-IR can alter glycemic control. In fact, the insulin receptor and IGF-IR form hybrid receptor complexes, and their molecular interactions have been characterized. 124,125 Hyperglycemia has been recognized as an associated AE related to treatment for TAO with teprotumumab. 111,112 This has been found, not only in the formal clinical trials but also in real-world experience. For instance, 52% of patients (22/42) developed hyperglycemia in one case series during treatment. 126 Others reported single patients developing severe hyperglycemia, 127,128 a hyperglycemic hyperosmolar state, 129 and a case of rapidly developing diabetic ketoacidosis, 130 none of which was observed in the phase 2 or phase 3 trials.
The IGF pathway plays important roles in the development and vitality of the ear and hearing. 131 Cochlear hair cell survival and maintenance are dependent on IGF-1 as is eustachian tube function. 121 Regarding hearing abnormalities identified in patients treated with teprotumumab, a spate of single case reports and case series has appeared, most commonly related to the frequency and persistence of these hearing changes/loss in a subset of patients. Among the first, Belinsky et al. reported 4 patients undergoing audiologic testing during/post-treatment developing hearing loss. 132 Unfortunately, none of these cases had undergone formal hearing testing at baseline. Importance of assessing hearing before treatment initiation is especially critical because of the relatively high incidence of audiologic changes in patients with GD and TAO. 120,121 This report was followed by several other case reports and case series, the majority of which included patients who had not been formally assessed both at baseline and during/after treatment. 133 –136 In a larger cohort, Sears et al. found that 81.5% (22/27) treated patients reported subjective hearing changes. 137 These reports are inherently limited by their near-complete reliance on subjective, nonstandardized patient questioning and reporting, thus making interpretation of the observations and results difficult. 138 This is evidenced by the subsequent study by Douglas et al., 139 which incorporated audiometry as a per protocol requirement, thus more objectively determining the actual incidence and severity of putative drug effects on hearing. That prospective study included 52 patients with TAO, each receiving 8 infusions of teprotumumab who had undergone both baseline and post-treatment audiometry, including at least a 6-month follow-up evaluation. Thirty-two out of 52 patients (mean age 43 years) had normal baseline audiometry. Following treatment, one of those patients with normal baseline studies (3%) developed grade 1 (mild) unilateral audiometric abnormality de novo. In contrast, baseline audiometry revealed abnormalities in 20/52 (38%) of patients (mean age 57 years). Of these, 10/20 (50%) developed a significant mild (grade 1) hearing decrease. With regard to subjective assessment, 6/52 (12%) of patients reported hearing symptoms at baseline. Tinnitus was a complaint of two patients, 3 reported bilateral hearing loss, and a single patient reported bilateral hyperacusis. During treatment, 15/52 (29%) patients reported new onset hearing symptoms not resolving at week 24 (end of therapy). At the 6-month follow-up, 11/15 patients had complete symptom resolution. Another recent report suggested that 11/22 patients manifested ototoxicity in at least one ear following treatment with teprotumumab. 140 The variable intervals of audiological testing used make definitive evaluation of the results difficult. In sum, audiometry and more subjective assessments based on patient reporting suggest that teprotumumab can alter hearing, more frequently in a subset of at-risk patients (older age, preexisting hearing loss, exposure to ototoxic sounds and drugs) and especially at higher frequencies. The weight of current evidence strongly supports prescriber diligence in obtaining audiometric assessment at baseline and during/following treatment with teprotumumab and apprising candidate patients of the risks for hearing loss from the drug treatment. 121
In sum, a number of gaps remain in our understanding, not only regarding the molecular and cellular biology of IGF-IR in its involvement with the development and clinical course of TAO but also in how we might better optimize its inhibition as a targeted therapeutic strategy for the disease. The postapproval ongoing clinical trials and those that will follow should provide even better refinements in our approach to managing this vexing disease.
Conclusions
TAO, associated most frequently with autoimmune diseases such as GD and Hashimoto’s thyroiditis, represents a vexing, potentially blinding disease process, which manifests in diverse clinical patterns. Historical perceptions regarding the pathogenesis of this disease and how it should be managed optimally are now yielding to evidence-based concepts that have directly resulted in specific, targeted medical treatments. A complex set of interactions between TSHR and IGF-IR appear to drive the phenotype of CD34+ fibrocytes, GD-OF, Tc, and Bc, both directly and indirectly. Identifying this molecular interplay has led to a targeted therapy now in common clinical use throughout the United States. Despite these substantial advancements, our current understanding of TAO and how to best manage it remain dwarfed by uncertainty. Ultimately, it is likely that newer and better-informed mechanistic concepts will result in even better therapies. Thus, we should feel optimistic that improved therapies for TAO, both medical and surgical, are in the offing.
Footnotes
Author’s Contributions
T.J.S. performed literature searches and wrote the article.
Author Disclosure Statement
T.J.S. was issued patents for targeting IGF-IR as therapy for autoimmune diseases, including TAO, held by Lundquist Institute/UCLA and University of Michigan; Consultant, Amgen (formerly Horizon), Viridian Therapeutics, Lundbeck, Argenx, Lassen Therapeutics, Minghui Pharmaceuticals, Bristol Myers Squibb, Acelyrin, Arrowhead Pharma, and Escient Pharma.
Funding Information
None beyond institutional support.