
Abstract
Background:
Thyroid cancer is the most common endocrine malignancy, with papillary thyroid cancer (PTC) accounting for ∼80% of all cases. DNA methylation alterations and gene expression changes in cancer, offer valuable insights into tumor biology and serve as potential clinical biomarkers. However, the functional implications of DNA methylation changes in PTC patients, particularly based on multiomics analysis in the Chinese population, remain insufficiently explored. This study aims to investigate the epigenetic signatures of thyroid cancer and identify the DNA methylation biomarkers for diagnosing PTC in Chinese patients.
Methods:
Thyroid cancer tissues (n = 40) and benign thyroid nodule tissues (n = 31) were collected from Chinese patients for global DNA methylation analysis. Gene expression profiles and H3K27ac modifications were also investigated to understand the impacts of epigenetic changes on gene expression. Genome-wide methylation profiling and machine learning methods were employed to differentiate PTC from control samples.
Results:
Genome-wide DNA methylation maps revealed significant methylome changes in Chinese PTC tissues. By integrating our data with The Cancer Genome Atlas thyroid cancer methylation profiles, we identified unique hypomethylation patterns associated with thyroid hormone synthesis specifically in Chinese PTC patients. RNA sequencing and H3K27ac modification analysis, along with functional assays, showed that the dysregulated genes in PTC patients are critical for the proliferation, migration, and invasion of thyroid cancer.
Conclusions:
Our study provides a comprehensive view of the multi-omics-based, function-guided DNA methylation landscape for Chinese PTC patients. We identified seven functional differentially methylated regions with high sensitivity and specificity for diagnosing thyroid cancer in Chinese patients. Additionally, DHRS3 is highlighted as a key player in PTC pathogenesis and shows promise as a valuable biomarker for predicting patient outcomes. This research advances our understanding of DNA methylation in thyroid cancer and underscores the importance of developing population-specific diagnostic tools to improve patient outcomes. However, further validation in larger, independent cohorts is needed to confirm their diagnostic value.
Introduction
Thyroid cancer is the most common endocrine malignancy. 1 Thyroid nodules are detected in up to 67% of the general population, 2,3 with 10–15% found to be malignant. 4 –8 Suspicious nodules identified by ultrasounds are typically examined further through fine-needle aspiration (FNA) and cytology, which usually classifies them as benign or malignant. However, cytology is not infrequently indeterminate so patients may require repeat FNA or additional testing. 9 –11 Over the past decade, molecular testing has been developed to clarify indeterminate cases and reduce unnecessary surgeries. 12 However, most of these diagnostic platforms have been developed and validated primarily in Western populations, raising concerns about their effectiveness in Chinese papillary thyroid cancer (PTC) patients. This highlights the need for a more accurate, population-specific diagnostic tool for thyroid nodules in China.
DNA methylation, a common epigenetic modification, plays a significant role in gene expression regulation. 13 Hypermethylation of gene promoter regions is frequently associated with the loss of gene function. Aberrant methylation patterns—characterized by hypermethylation of tumor suppressor genes and hypomethylation of oncogenes—are hallmark features of many human cancers. 14 –17 These epigenetic alterations disrupt essential cellular processes, including transcription, DNA repair, and replication, thereby contributing to tumor initiation and progression. 16 –20 Notably, DNA methylation changes have already been established as effective biomarkers for the early detection and prognosis of various cancers. In thyroid cancer, Yim et al. identified 373 DNA differential methylation sites distinguishing PTC from benign nodules. 21 Recently, Hong et al. refined this approach by identifying key DNA methylation biomarkers for cell-free DNA and creating a model based on the most statistically significant features. 22
Despite these advances, existing models are often limited by overfitting due to small sample sizes and the heavy reliance on statistical algorithms. Additionally, differences in methylation patterns across populations emphasize the need for population-specific research. 23,24 To address this, we conducted a comprehensive analysis of DNA methylation patterns in Chinese thyroid cancer patients by integrating multiomics data with function-guided biomarker selection.
Specifically, we integrated DNA methylation data generated by reduced representation bisulfite sequencing (RRBS), H3K27ac signals from chromatin immunoprecipitation sequencing (ChIP-seq), and RNA expression data from RNA sequencing (RNA-seq) to characterize the potential functional DNA methylation biomarkers. ChIP-seq, which combines chromatin immunoprecipitation with high-throughput sequencing, enables the identification of DNA-associated proteins across the entire genome, thereby defining key regulatory elements. 25,26 Similarly, RNA-seq quantifies RNA levels and sequences, providing insights into gene expression patterns. 27 To explore population-specific epigenetic features, we compared our methylation data with that of The Cancer Genome Atlas (TCGA) patient samples, 28 which utilizes Illumina 450K array to profile methylation in PTC versus adjacent thyroid tissues. 29 Using machine learning approaches, we identified and validated selected differentially methylated region (DMR) candidates through functional assays to assess their biological significance.
Materials and Methods
Human tissue collection and pathological confirmation
Thyroid tumor tissues and benign tissues were collected from patients undergoing thyroid surgery at the Department of Thyroid and Neck, Tianjin Medical University Cancer Institute and Hospital from July 2020 to July 2021. All samples were retained with patients’ informed consent for use in this study. Detailed patient and sample information was provided in Supplementary Table S1. All protocols, including informed consent, were approved by the Tianjin Medical University Cancer Institute and Hospital Ethics Committee (EK2021149).
Patient population
A total of 90 patients were retrospectively selected and categorized into developmental and validation groups. Sixty tissue samples were assigned to the developmental group, with 11 samples excluded due to insufficient material or incomplete clinical information, leaving 30 PTC and 19 benign nodules for analysis. Among these, three samples with sufficient material were selected for RNA-seq and H3K27ac ChIP-seq. In the validation group, 30 tissue samples were initially assigned, with 8 samples excluded due to insufficient material, incomplete clinical information, or unsuccessful RRBS library construction, 10 PTC and 12 benign nodules remained for analysis (Fig. 1 and Table 1).

Study design flow diagram. Schematic representation of patient selection, detailing inclusion, and exclusion criteria for both the developmental and validation cohorts. BTN, benign thyroid nodule; ChIP-seq, chromatin immunoprecipitation sequencing; PTC, papillary thyroid carcinoma; RNA-seq, RNA sequencing; RRBS, reduced representation bisulfite sequencing.
The Clinical Characteristics of Chinese Patients in This Study
All ultrasound examinations used a Phillips EPIQ 5, IU 22, HD11 (Philips Healthcare, Eindhoven, The Netherlands) equipped with a high-frequency linear array probe (5–12 MHz) to perform thyroid cross-section, longitudinal section, and cervical lymph node scan. The molecular events (BRAF mutation) were characterized with a 177-gene panel that covering all the hotspots from COSMIC database.
CLNM, central lymph node metastases; DDMS, Diagnostic DNA Methylation Signature approach; LLNM, lateral lymph node metastases; RLN, recurrent laryngeal nerve; TNM, tumor-node-metastasis classification: Tumor staging (T/N/M) was classified according to the 8th edition of the American Joint Committee on Cancer (AJCC-8) staging system.
DNA isolation and RRBS library preparation
Genomic DNA was isolated from fresh frozen tissue samples using Magnetic Universal Genomic DNA Kit (TianGen, China). A total of 1.5 µg of genomic DNA, spiked with lambda DNA, was digested with MspI. Bisulfite conversion was performed using the EZ DNA Methylation-GoldTM Kit (Zymo Research, USA). The quality of the prepared RRBS libraries was examined using Agilent Bioanalyzer 2100 system. A detailed library construction protocol was provided in the Supplementary Data.
RRBS data analysis
The RRBS libraries were sequenced on the Illumina NovaSeq platform. Bismark software (version 0.16.3) was used to align to the reference genome (hg38). 30 The R package methylKit (v1.32.0) was employed to perform differential DNA methylation site analysis. 31 To ensure high-quality data, the DNA methylation sites with low coverage (less than 10 reads) were filtered out. A cutoff of 0.1 for the absolute change in methylation levels was set to define differential DNA methylation CpG sites (DMCs) with p value ≤0.01. The read counts and mapping ratios were provided in Supplementary Table S1.
ChIP-seq data analysis
H3K27ac ChIP-seq sequencing reads were aligned to the genome (hg38) using Bowtie2 (v2.2.5). Peak calling was performed using MACS (v2.1.0). 32 The bdgdiff model of MACS software was applied to identify differential peaks between two conditions. The H3K27ac modification profile around enhancer regions was analyzed using deepTools (v3.5.5). 33 The results generated by MACS were loaded into IGV (v2.8.13) for visualization. 34
RNA sequencing data analysis
The RNA sequencing data were analyzed using the open-source software package RNACocktail (v0.3.2). 35 Quality control was conducted with FastQC (v0.11.9) and Trimmomatic (v0.39). 36 HISAT2 (v2.2.1) 37 was used to align the qualified reads to the reference genome (hg38). The generated BAM files were sorted and indexed by SAMtools (v0.1.19). Stringtie (v2.0) was used for transcript assembly and annotation. 38 The DESeq2 (v1.30.1) was used for differential expression analysis, 39 using a significance threshold of p ≤ 0.05 and |log2-fold change| ≥ 1.
Classification model development
The least absolute shrinkage and selection operator (LASSO) logistic regression method was built using glmnet package. To minimize model overfitting, we focused on DMRs associated with 212 hypomethylated-upregulated and 30 hypermethylated-downregulated genes in PTC tissues. Through 10-fold cross-validation, seven DMRs were identified as key features (Supplementary Table S2).
Cell culture, transfection, and quantitative real-time PCR
K1 and HEK293T cell lines were purchased from American Type Culture Collection (ATCC) and cultured in RPMI-1640 and DMEM (Gibco, USA) supplemented with 10% fetal bovine serum. To knock down DHRS3, ADM and RIN1, three RNAi oligos specific to each gene were synthesized by Sangon Biotech and transfected into cells using RNAiMAX (Invitrogen, USA). Total RNA was extracted using TRIzol reagent (SparkZol, China). Quantitative real-time PCR (qPCR) was performed using SYBR Premix Ex Taq II (Roche, Switzerland). Primers were listed in Supplementary Table S3.
Transwell migration and invasion assays
Cell migration and invasion assays were performed using a polycarbonate membrane transwell chamber (Corning, USA) in 24-well plates. 40 For cell migration assay, 100 µL of cell suspension (1 × 106 cells/mL) in serum-free medium was added to the upper chamber. For cell invasion assay, transwell polycarbonate membranes were precoated with Matrigel (Corning, USA), and then cells were added on top of Matrigel coating upper chamber. For a detailed protocol, please refer to the Supplementary Data.
Statistical analysis
All statistical analyses were performed using R software (v4.3.2). The Wilcoxon rank-sum test was used to assess significant differences in DNA methylation levels between the two groups, with a cutoff value of 0.1 and p value ≤0.01 considered statistically significant. Wilcoxon’s signed-rank test was applied to evaluate significant differences in differential expression, with p values <0.05 considered significant. Receiver operating characteristic (ROC) curves were used to estimate sensitivity, specificity, and the area under the curve (AUC). For experimental validation, GraphPad Prism (v10.1.1) was used for data visualization and significance testing. Data are presented as mean ± SD, with the number of replicates and statistical tests described in the figure legends for each panel.
Results
Genome-wide DNA methylation changes in Chinese thyroid cancer patients
To explore the utility of methylation patterns in differentiating papillary thyroid cancers, we collected and analyzed the whole-genome methylation profiles of thyroid specimens from Chinese PTC (n = 40) and benign thyroid nodule (BTN) tissues (n = 31) (Figs. 1 and 2A, Table 1). We found that 172,931 CpG sites (3.2%) were hypomethylated in PTC, while 51,169 CpG sites (0.9%) showed increased methylation (Supplementary Fig. S1A). Next, we focused on the DMRs between PTC and BTN. By categorizing these clustered regions into lowly (<0.2), medium (0.2–0.8), and highly (>0.8) methylated levels, we found a reduced proportion of highly methylated regions in PTC tissues (Fig. 2B). Using a ≥0.1 difference cutoff, we identified 5216 hypomethylated and 1441 hypermethylated DMRs (Supplementary Fig. S1B), highlighting numerous methylation alterations in PTC tissues. Additionally, the identified DMRs had a mean size of 226 bp, suggesting the specific larger regions are altered (Fig. 2C, Supplementary Fig. S1C). Across DMRs, there was a clear trend toward reduced methylation in PTC (Fig. 2D, E), and these methylation differences allowed us to distinguish PTC from BTN tissues (Fig. 2F, Supplementary Fig. S1D). Overall, the distinct methylation profiles between Chinese PTC and BTN samples were widely distributed across all chromosomes (Supplementary Fig. S1E), indicating that genome-wide epigenetic alterations occur during PTC progression.
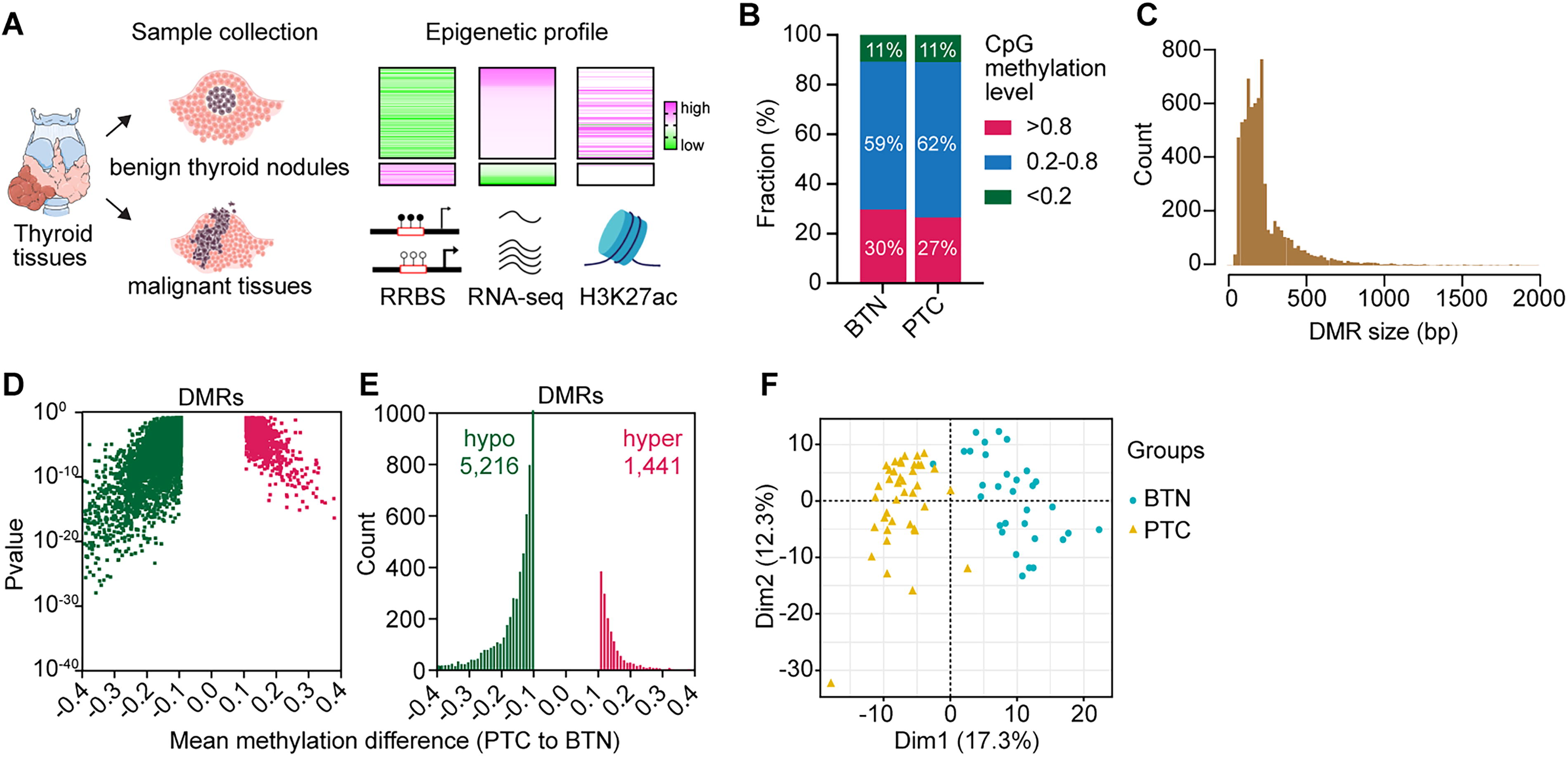
The DNA methylation landscape of Chinese thyroid cancer. (
DNA methylation changes in regulatory regions of Chinese thyroid cancer patients
To understand the functional impact of these methylation changes, we linked DMRs to their nearest genes. In total, we identified 3599 genes with at least one DMR located within 5 Kb upstream of the transcription start site (TSS) to 5 Kb downstream of the transcription termination site (TTS) (Fig. 3A, B). Analysis revealed that DMRs exhibiting DNA methylation differences greater than 0.1 were significantly associated with disease status, specifically distinguishing PTC from BTN samples. However, no significant differences were observed based on sex (male, n = 22 vs. female, n = 49) nor age (age ≤50 years, n = 41 vs. age >50 years, n = 30) (Fig. 3C). Notably, ∼90% of the hypomethylated DMRs and 47% hypermethylated DMRs are located near enhancer regions (Fig. 3D, E, and Supplementary Fig. S2A). Gene ontology analysis revealed that DMR-associated genes (PTC vs. BTN) were related to cancer development and progression (Supplementary Fig. S2B, C).
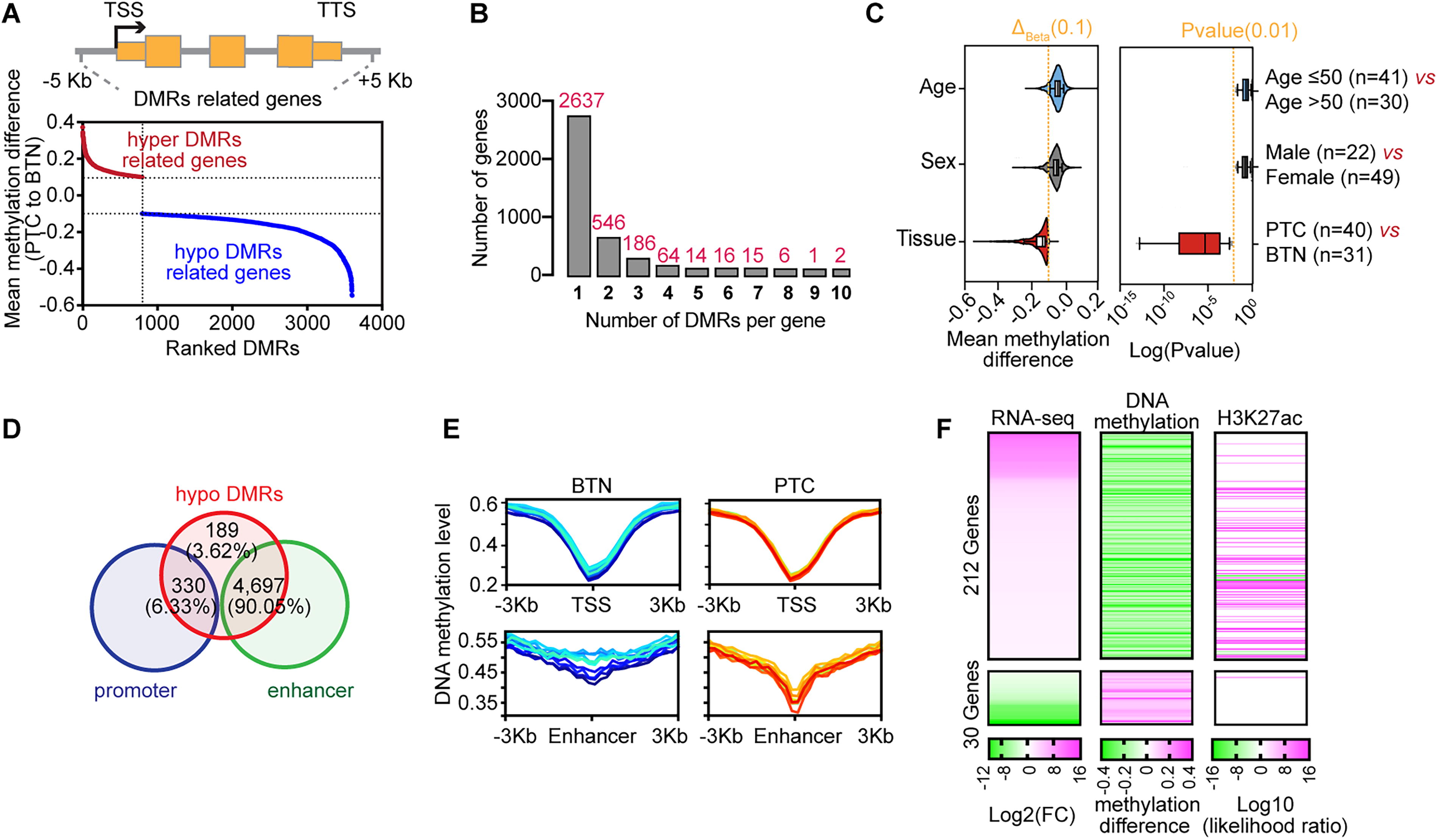
DNA methylation and gene expression changes in Chinese PTC patient samples. (
To further explore the relationship between gene expression and DNA methylation in PTC tissues, we performed RNA-seq and H3K27ac ChIP-seq on PTC and paired adjacent tissues (Fig. 2A). By integrating DNA methylation and expression data, we classified DMR-associated genes (PTC vs. BTN) into four groups (Supplementary Fig. S2D). In PTC tissues, 212 genes were concurrently hypomethylated and upregulated, while 30 genes were hypermethylated and downregulated (Fig. 3F, Supplementary Fig. S2D, E). Interestingly, these hypomethylated DMRs were also broadly marked by H3K27ac, suggesting that the DNA methylome aligns with enhancer activity to regulate gene expression in PTC (Fig. 3F, Supplementary Fig. S2F). Overall, the methylation changes, especially hypomethylation in regulatory regions, play a key role in gene expression regulation in Chinese PTC patients.
Hypomethylated thyroid hormone synthesis pathway in Chinese PTC patients
To identify the DNA methylation changes specifically to Chinese PTC patients, we analyzed Illumina 450K DNA methylation data for thyroid carcinoma (THCA) from TCGA 29 (Supplementary Fig. S3A). When integrating these data with our RRBS dataset, we focused on common observations, such as overlapping methylated sites and regions, to capture biologically relevant signals that are less influenced by technical variation. In the TCGA patient samples (PTC n = 507 vs. adjacent normal thyroid tissues [NT] n = 56), we identified 15,346 differentially methylated CpG sites (DMCs) (Fig. 4A). Among these DMCs, 575 were hypomethylated and 98 were hypermethylated, showing the same patterns in our RRBS data (Fig. 4B, Supplementary Fig. S3B). However, 525 CpG sites exhibited opposite methylation patterns between the two datasets (450K from TCGA vs. our RRBS; Fig. 4B, bottom panel).

DNA methylation patterns specific to thyroid cancer in the Chinese population. (
By clustering the 450K CpG sites into regions and identifying DMR-associated genes, we found that the differentially methylated genes, identified in the Chinese patients compared to the TCGA patient population, were enriched in cancer-related pathways, including thyroid hormone synthesis (Fig. 4C, D). Thyroid hormones not only regulate the physiological processes of normal cells but also stimulate cancer cell proliferation via dysregulation of molecular and signaling pathways. 41 –44 Notably, hypomethylated genes specific to Chinese PTC patients in this pathway include ASGR1, DUOX1, DUOXA1, DUOX2, and DUOXA2 (Fig. 4E–G, Supplementary Fig. S3C–F), and these changes were accompanied by increased H3K27ac modifications (Fig. 4E–G, Supplementary Fig. S3G). These hypomethylation patterns were further validated by methylation-specific PCR assays (Supplementary Fig. S3H). DUOX1 and DUOX2 encode enzymes responsible for generating hydrogen peroxide (H2O2), which is essential for thyroid peroxidase-mediated iodination reactions in normal thyroid hormone synthesis. 45,46 The upregulation of DUOX1/2 may contribute to an increased oxidative environment within cancerous thyroid tissue, potentially promoting DNA damage and driving tumorigenesis. 47,48 Overall, our findings indicated that DNA methylation changes in PTC vary significantly between populations.
Integrative data analysis reveals epigenetically regulated genes in PTC
Integration of our RRBS data with RNA-seq analysis revealed that 212 genes were hypo-upregulated in Chinese PTC patients compared to BTN tissues (Figs. 5A and 3F). Notably, 95 of them exhibited the same regulatory patterns in the TCGA patients (Fig. 5B, C). In contrast, 30 genes were hyper-downregulated in PTC tissues from the Chinese population, with 8 of them overlapping with the TCGA patient samples (Fig. 5B, D). We also performed H3K27ac ChIP-seq analysis on Chinese PTC patients to access enhancer-associated epigenetic changes. Integrating RRBS with RNA-seq and H3K27ac ChIP-seq, we found that DHRS3, ADM, and RIN1 exhibited the most significant and consistent alterations in DNA methylation (Supplementary Fig. S4A–G), histone modifications, and gene expression across our datasets (Fig. 5E–G, Supplementary Fig. S4H–I). Among the common hypo-upregulated genes, DHRS3 consistently identified in the Chinese and TCGA PTC patients (Fig. 5E, Supplementary Fig. S5A, B). High expression levels of DHRS3 correlated with poorer survival probabilities in the TCGA dataset, supporting the robustness of our analysis and highlighting its potential as a prognostic marker (Supplementary Fig. S5C). Our RRBS data further revealed three hypomethylated DMRs associated with upregulation of DHRS3 in PTC tissues (Fig. 5F, Supplementary Fig. S5D, E). Additionally, ADM and RIN1 were also found to be hypo-upregulated in both populations (Fig. 5E). ADM has been recognized as a prognostic biomarker for thyroid cancer since 2022, 49 while RIN1 exhibited the most significant methylation changes and the greatest difference in gene expression between Chinese PTC and BTN tissues (Fig. 5A, E). The hypomethylation and upregulation of DHRS3, ADM, and RIN1 were further validated by MSP assays (Supplementary Fig. S6A) and independent RNA-seq data from Chinese PTC patients (HRA000779) 50 (Fig. 5H and Supplementary Fig. S6B–D). Additionally, the expression patterns of these three genes were linked to disease progression, including T stage, lymph node metastasis, and the overall clinical stage of thyroid cancer (Supplementary Fig. S6E–H).
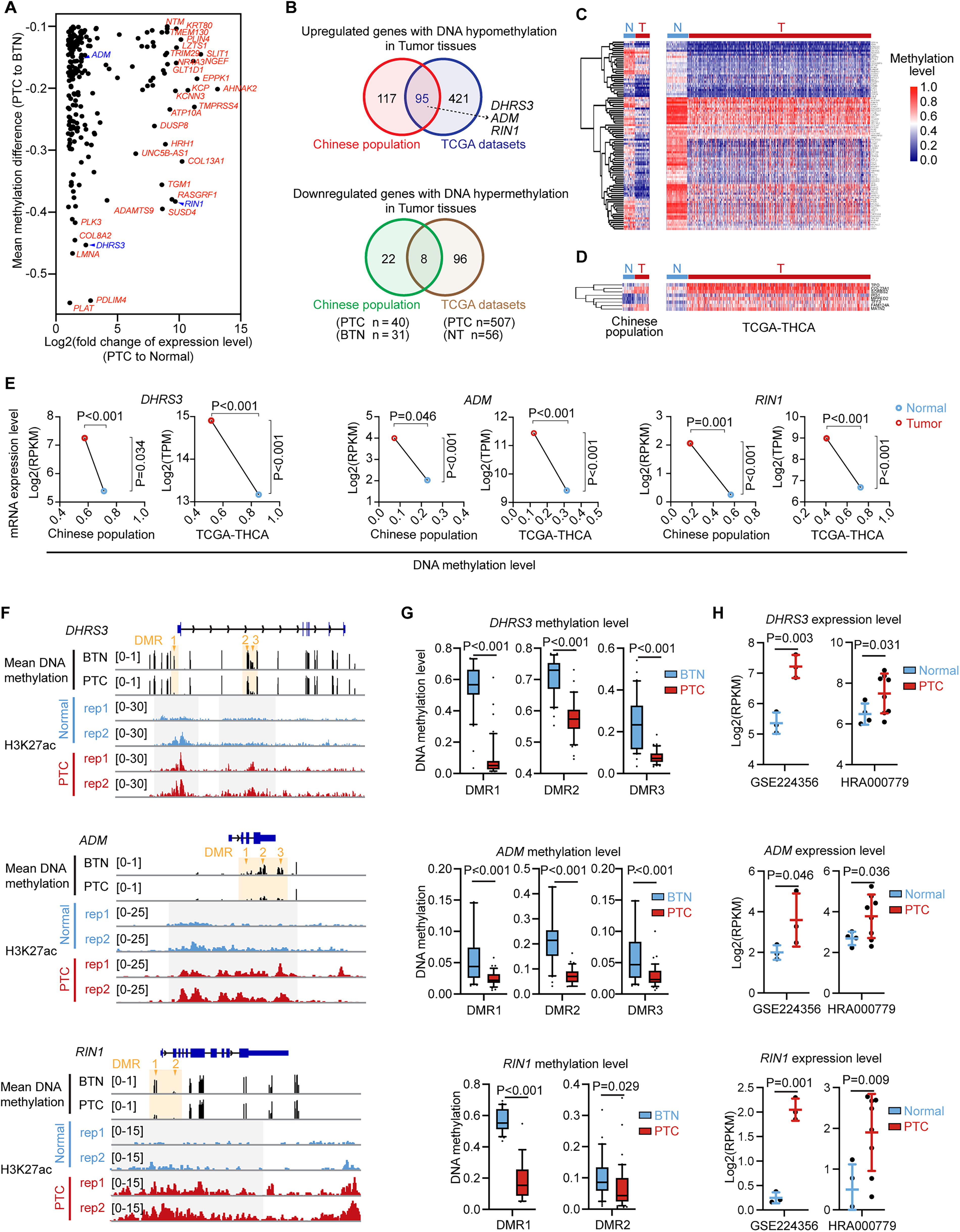
Integrative analysis reveals the epigenetically regulated genes in thyroid cancer. (
The functional role of DMR-associated genes in thyroid cancer
To investigate the functional roles of these three candidates in PTC, gene knockdown was carried out in the K1-cell line (Fig. 6A). Disruption of any of the three genes significantly suppressed cell proliferation, as demonstrated by cell viability and colony formation assays (Fig. 6B–D). Additionally, knockdown of three genes dramatically reduced cell migration and invasion (Fig. 6E, F). To further examine the regulatory role of DMR-associated enhancers, we performed CRISPR interference (CRISPRi) experiments by targeting sgRNAs to the enhancer regions of DHRS3 and ADM (Fig. 6G). We found that inactivating these DMR-related enhancers led to a significant repression of their gene expression (Fig. 6H). In summary, our finding suggested that DNA methylation alterations, along with other epigenetic changes such as H3K27ac modifications, regulate cell proliferation, migration, and invasion through functional elements and their associated genes, thereby promoting thyroid cancer development.
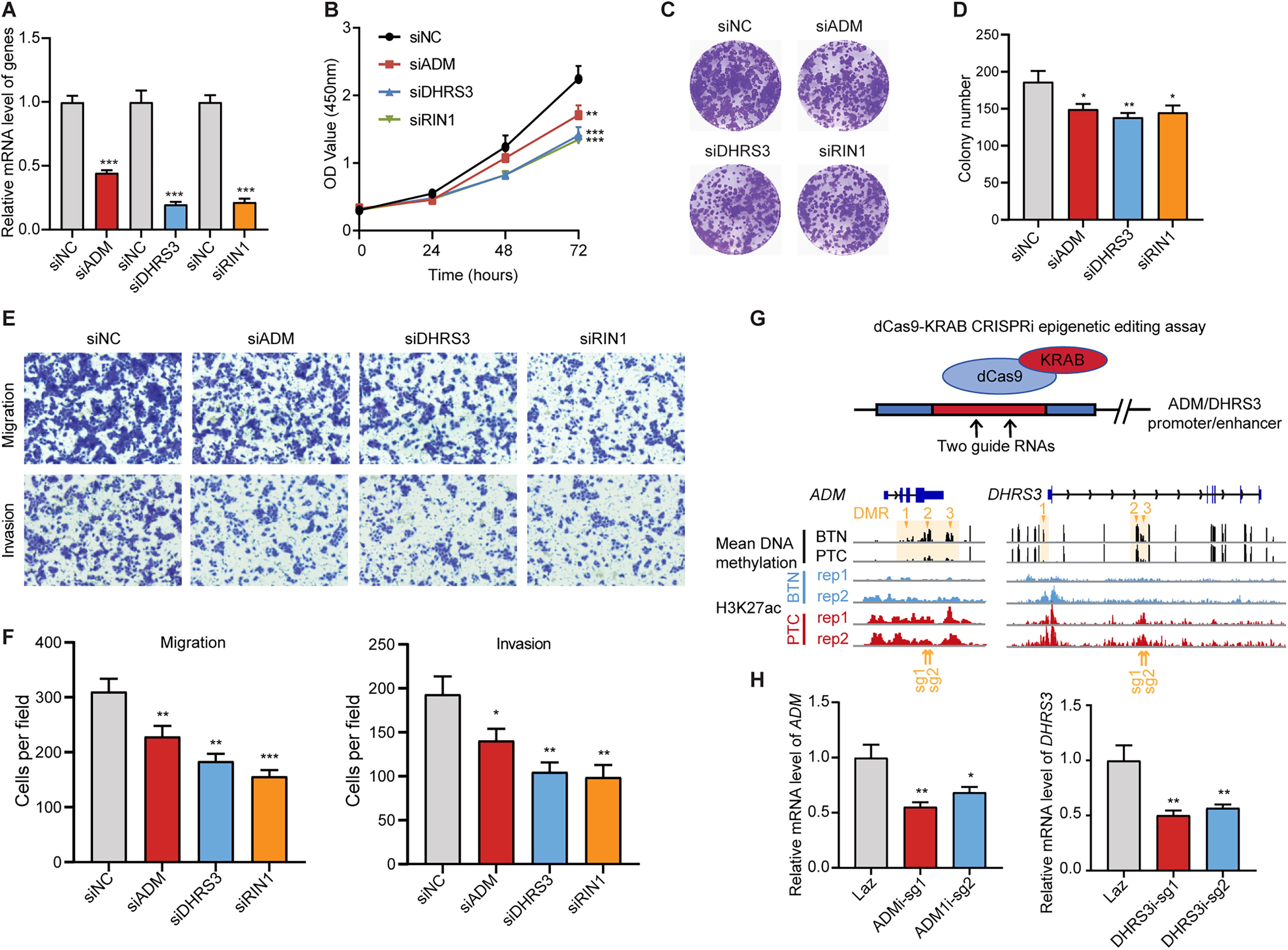
The hypomethylated genes in thyroid cancer regulate cell proliferation and metastasis.
Methylation profiles predict diagnosis of thyroid cancer
To explore the utility of methylation patterns in thyroid cancer and improve diagnosis, we used a LASSO formula to select key PTC methylation features from DMRs associated with 212 hypo-upregulated genes and 30 hyperdownregulated genes identified in Chinese PTC patients (Fig. 3F, Supplementary Fig. S2D). Based on the 10-fold cross-validation using the training patient samples, seven DMRs were selected, with six being hypomethylated and one were hypermethylated in the Chinese PTC patients (Fig. 7A, Supplementary Fig. S7A, B, and Supplementary Table S2).
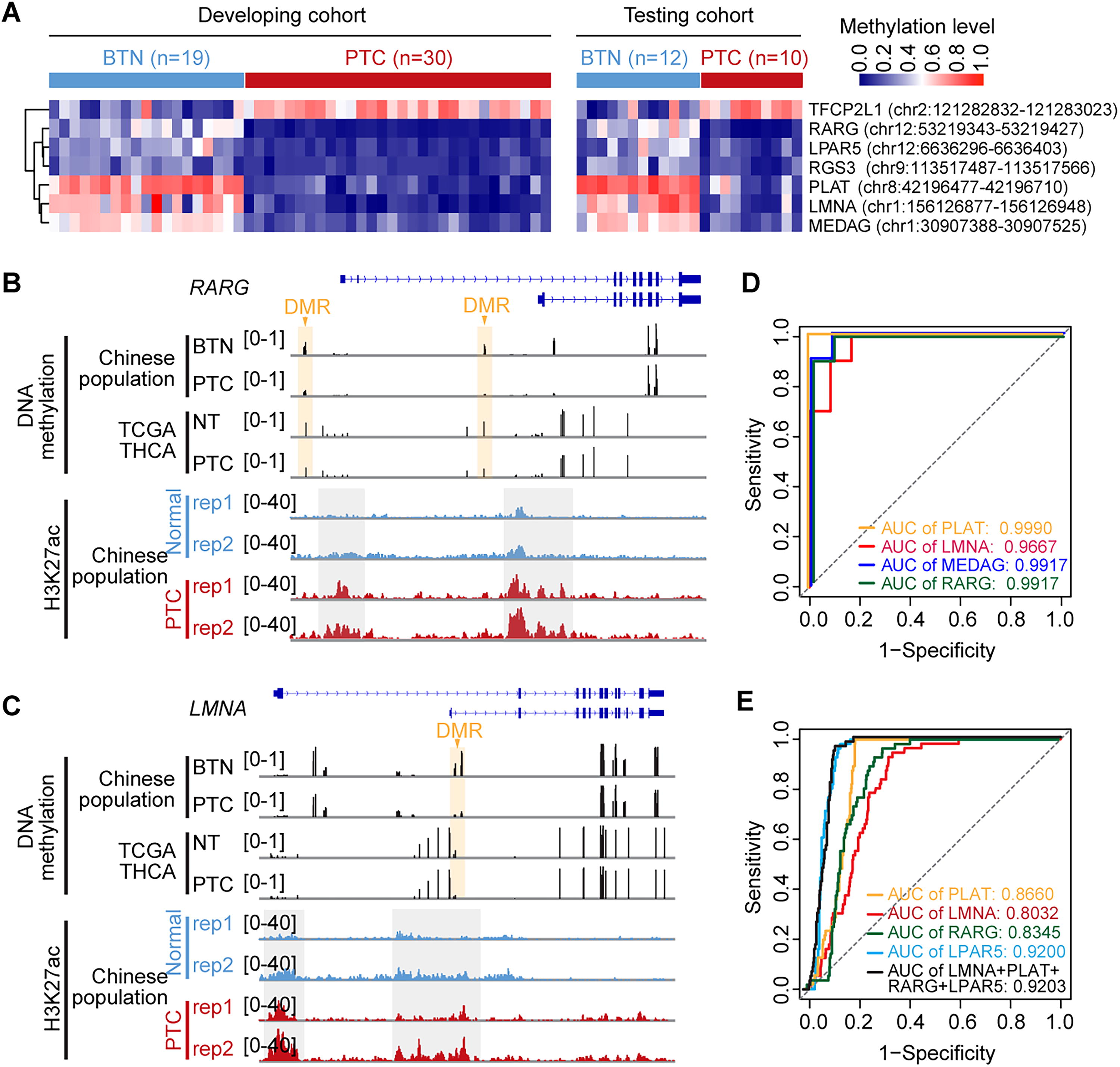
DNA methylation marker selection and validation for Chinese PTC patients. (
Among these seven identified DMRs, four showed consistent methylation patterns in the TCGA patient samples, demonstrating their relevance across different populations (Fig. 7B, C, Supplementary Fig. S7C). The diagnostic accuracy of individual DMRs for Chinese PTC was demonstrated with an AUC exceeding 0.99 in testing patient samples (Fig. 7D), and they also demonstrated strong diagnostic accuracy in the TCGA dataset, further validating their potential as universal biomarkers for PTC (Fig. 7E). The remaining three DMRs may serve as specific diagnostic markers for the Chinese population. For example, the MEDAG gene was hypomethylated only in Chinese PTC patients, but not in the TCGA patient population (Supplementary Fig. S7D). Overall, our results underscore the reliability of methylation patterns in identifying thyroid cancer and highlight specific signatures unique to Chinese PTC samples.
Discussion
The increasing prevalence and incidence of papillary THCA has made it a significant global health concern. Although PTC is often an indolent disease associated with excellent overall survival rates, accurate and early diagnosis is critical. DNA methylation often alters in cancer cells, leading to changes in gene expression that can contribute to tumor development and progression. 51 Tumor-specific methylation patterns have also been extensively studied for their potential applications in cancer diagnosis and prognosis. 52,53 In this study, we identified global methylation changes specific to Chinese PTC patients and developed a DNA methylation marker-based model for thyroid cancer diagnosis.
Most studies in DNA methylation biomarker discovery rely primarily on mathematical statistics, often overlooking the genomic context and biological relevance of the identified markers. To overcome this limitation, we developed a function-guided selection strategy based on multiomics integration. Specifically, we combined DNA methylation data with H3K27ac signals and RNA expression profiles to identify functional biomarkers that could distinguish malignant thyroid nodules from benign ones. With this logic, we prioritized the DMRs that were located within the functional genomic elements and affected nearby gene expression as biomarker candidates, increasing the likelihood of selecting biologically meaningful markers.
In total, we identified 5216 hypomethylated and 1441 hypermethylated DMRs, with 212 genes exhibiting hypomethylation accompanied by upregulation in Chinese PTC tissues. To validate our strategy, we selected the top-ranked genes—DHRS3, ADM, and RIN1—as examples for validation. These genes exhibited consistent DNA methylation changes across multiple validation platforms, including MSP, ChIP-seq, and RNA-seq, highlighting their potential functional relevance in thyroid cancer. Functional assays further confirmed that these genes play crucial roles in tumor development and progression, including cell proliferation, migration, and invasion. Notably, DHRS3 emerged as a potential prognostic marker for PTC, underscoring its utility in predicting disease outcomes and guiding clinical management. Consistent with our findings, ADM has previously been identified as a prognostic factor in thyroid cancer. 49 Additionally, ADM, in combination with PXDN, MMP1, and TFF3, has been characterized as a diagnostic biomarker for anaplastic thyroid cancer, with an AUC >0.75 in ROC analysis. 54 Through LASSO regression and ROC analyses, we further demonstrated that the methylation patterns of seven specific DMRs could serve as highly sensitive and specific biomarkers for the accurate diagnosis of PTC in the Chinese population.
An important aspect of our study is the integration of data derived from different methodological platforms to characterize DNA methylation changes specific to the Chinese PTC patient population. The Chinese patients were analyzed using RRBS, a sequencing-based approach that offers high resolution in CpG-dense regions, while the TCGA dataset was generated using the Illumina 450K array, a microarray-based method that interrogates a predefined set of CpG sites across the genome. Although both methods accurately represent DNA methylation levels, they differ in coverage, resolution, and technical biases. Our study provides a much higher resolution of previously unexplored regions (e.g., enhancers), significantly extending and revealing pronounced genome-wide DNA methylation and gene expression changes in Chinese PTC patients. To address methodological differences, we focused on overlapping methylated sites and DMRs that were consistently identified in both datasets. This approach allowed us to capture biologically relevant signals while minimizing the influence of technical variation.
Several limitations of this study should be acknowledged. First, our validation samples were relatively small, and further sample collection and validation are needed in future studies to enhance the robustness of our findings. Second, there were differences in control groups between the study populations, with PTC versus BTN in the Chinese patients and PTC versus adjacent tissue in the TCGA patient samples. The comparison of PTC versus BTN is highly relevant, as identifying methylation changes between these groups could improve the differential diagnosis of thyroid nodules, making it a key consideration in our study design. Last, while our multiomics approach successfully identified candidate biomarkers, additional experimental and clinical validation, including independent patient samples and functional assays, are needed to confirm their utility in routine clinical diagnostics. Currently, commercial molecular diagnostic products are primarily based on DNA, RNA, or microRNA. Integrating DMR biomarkers with existing DNA/RNA panels could enhance diagnostic accuracy, making it a promising direction for future research.
Conclusions
Our multiomics analysis of the DNA methylation landscape in Chinese PTC patients reveals key epigenetic differences in disease pathogenesis. Integrating DNA methylation with gene expression and H3K27ac data, we identified seven functional DMRs with high diagnostic accuracy and highlighted DHRS3 as a potential prognostic biomarker. These insights offer a foundation for enhancing thyroid cancer diagnosis and informing disease management in the Chinese population. Further confirmatory studies in larger and independent cohorts along with functional validation are essential to further establish their clinical utility.
Footnotes
Acknowledgment
The authors thank Min Zhao from the University of the Sunshine Coast for the draft and figure editing.
Authors’ Contributions
X.Z., M.Z., and M.G.: Conceptualization, funding acquisition, writing review, and editing. X.R.: Conceptualization, data curation, experiments and data analysis, funding acquisition, methodology, and writing original draft. F.Y.: Conceptualization, bioinformatics analysis, experiments and data analysis, funding acquisition, methodology, writing original draft. M.L. and Z.R.: Bioinformatics analysis. Q.D.: Experiments and data analysis. W.H., X.Y., J.Z., and X.Q.: Resources. W.Z.: Experiments and data analysis. D.L.: Resources and funding acquisition. T.G.: Writing original draft. L.W.: Data curation, bioinformatics analysis, and methodology. Y.P.: Data curation. All the authors have read and approved the final version of the article.
Author Disclosure Statement
The authors declare no potential conflicts of interest.
Data Availability
The raw and processed RRBS data were deposited to Gene Expression Omnibus (GEO) with accession number GSE233560. The RNA-seq data and H3K27ac ChIP-seq data were submitted to the GEO database under the accession ID GSE224357.
Funding Information
This work was sponsored by Shanghai Pujiang Program (21PJ1421800), The National Natural Science Foundation of China (82172821, 82103386, 82272721, 82471902, 32400431), Tianjin Municipal Science and Technology Project (19JCYBJC27400, 21JCZDJC00360) and Beijing-Tianjin-Hebei Basic Research Cooperation Project (20JCZXJC00120), The Science & Technology Development Fund of Tianjin Education Commission for Higher Education (2021ZD033), Tianjin Medical Key Discipline (Specialty) Construction Project (TJYXZDXK-058B), Tianjin Health Research Project (TJWJ2022XK024), and Talent Excellence Program from Tianjin Medical University (to M.Z.).
Supplementary Material
Supplementary Data
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.