
Abstract
Background:
Papillary thyroid carcinoma (PTC) is the most common malignant tumor of the endocrine system. BRAF mutations occur in 40–60%, panRAS mutations in 10–15%, and different gene fusion events such as RET fusions in 7–35% of these neoplasms. Artificial intelligence (AI) methods could be used to predict genetic changes from conventional histopathological slides.
Methods:
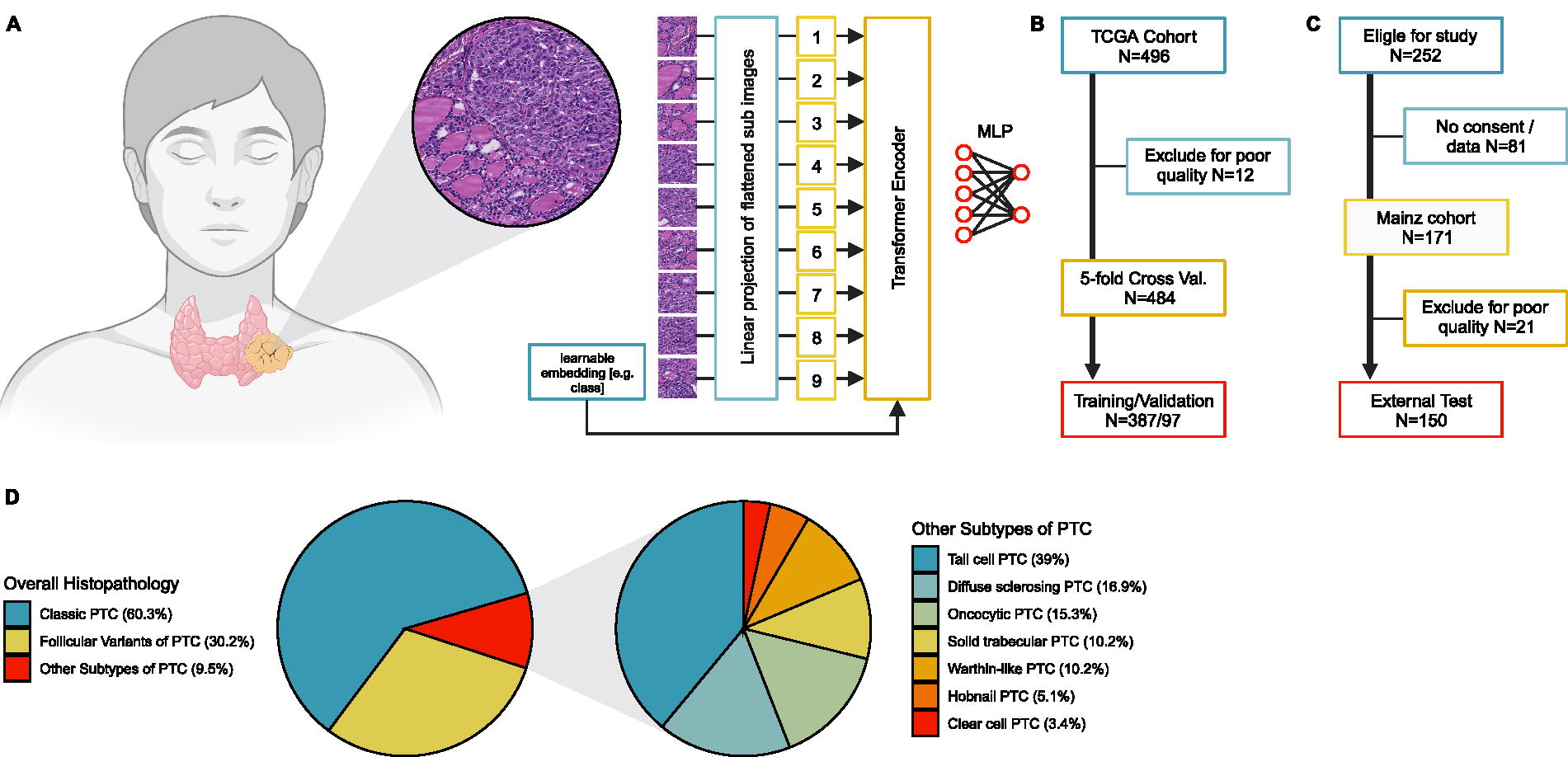
In this retrospective study, we used two independent cohorts of patients with PTC, totaling 662 cases for the establishment of our AI pipeline. The Cancer Genome Atlas cohort (496 cases) served as the developmental cohort, while the Mainz cohort (166 cases) served as an independent external test cohort. BRAF, panRAS, and fusion status was determined for all of these patients as target variables. Vision Transformer was trained on digitized annotated hematoxylin and eosin-stained slides for the presence of these alterations. Highest probability image tiles were used to identify new morphological criteria associated with the genetic changes.
Results:
The trained model resulted in an area under the receiver operating characteristic curve of 0.882 (confidence interval 0.829–0.931) for BRAF, 0.876 (0.822–0.927) for panRAS, and 0.858 (0.801–0.912) for gene fusions. Accuracy was 79.3% (72.7–85.8%) for BRAF, 89.3% (84.2–94.0%) for panRAS, and 84.7% (78.8–90.2%) for gene fusions. The performance on the validation set was almost identical to that on the test set. Analyzing the highest predictive tiles, novel morphological criteria for fusion-associated PTC could be discovered.
Conclusions:
Our study demonstrates that predicting genetic alterations in digitized histopathological slides using AI is feasible in patients with PTC. Our model showed high accuracy in predicting these changes, making it potentially suitable for pre-screening. Explainability approaches uncovered previously undescribed morphological patterns associated with certain genotypes. Providing pathologists with these AI-based features could improve their accuracy. Assuming further positive prospective validation, this discovery could contribute to a deeper understanding of PTC.
Introduction
Thyroid carcinomas are the most common endocrine malignancy. As of 2020, the mortality rates persist at a constant level of 0.5 per 100,000 person years in the United States. 1,2 Approximately 80% of thyroid carcinomas are diagnosed as papillary thyroid carcinoma (PTC), 3 which includes distinct subtypes characterized by different growth patterns, stromal changes, and cellular appearances. 4 BRAF mutations were reported in between 40% and 60%, 5,6 panRAS alterations in 10–15%, 6,7 and gene fusions, such as RET protooncogene fusions, in around 7–35% 6 of PTCs. Diagnostic techniques include sonography, fine needle aspiration (FNA), scintigraphy, computed tomography imaging or magnetic resonance imaging, but final diagnosis usually requires histopathology. 3,8 Additionally, molecular pathology plays an important role in contributing to differential diagnosis, prognosis determination, and the design of targeted therapies. Standard therapy protocols involve surgical intervention, thyroid hormone administration, and radioiodine therapy and is often influenced by different genetic alterations. There are also additional options of targeted therapy for patients with RET, 9,10 NTRK, 11 BRAF, 12 and panRAS alterations. 13 Early detection of these mutations is therefore an important part of optimizing therapy strategies and determining patient prognosis. 7,14 –16
Artificial intelligence (AI) holds enormous potential for a more efficient and cost-effective approach to mutation detection from conventional histopathology. 17 This could enhance the speed and affordability of diagnosing and treating PTC compared with traditional methods. However, the use of AI in predicting molecular alterations from histopathology in PTC remains relatively unexplored. Previous studies of Anand et al. 18 and Tsou et al. 19 have focused solely on BRAF/RAS axis alterations, neglecting other genomic targets. Newer algorithms, such as Vision Transformers (ViTs), offer a promising solution, demonstrating equivalent accuracy to traditional methods but with reduced computational demand and reduced image-specific bias. 20,21 Their robustness against adversarial attacks makes them more suitable for integration into clinical practice, ensuring better security of patient data. 22
Therefore, the aim of this study was to develop a reliable ViT-based deep learning model for predicting genomic alterations in PTC from histopathological slides. We also intended to visualize the classification results and potentially identify new morphological patterns associated with certain genomic alterations using explainability approaches. This could contribute to the advancement of AI methods in diagnosis and treatment stratification of PTC.
Materials and Methods
Patient cohorts
Two cohorts of patients with papillary thyroid cancer were used. The first cohort consisted of 496 patients of The Cancer Genome Atlas (TCGA)-THCA dataset diagnosed between 2000 and 2013. A detailed description can be found in the references. 6 All necessary information, including clinicopathological and sequencing data, were gathered from https://www.cbioportal.org/ as well as the Genomic Data Commons (GDC) data portal. Formalin-fixed paraffin-embedded (FFPE) tissue slides (in the context of the TCGA consortium referred to as diagnostic slides) were digitized using different whole slide scanners at the respective institutions. Slides from these patients were used to train the deep learning model and perform an initial five-fold cross-validation. The second cohort was generated at the University Medical Center in Mainz and consisted of 171 patients diagnosed with the same entity between 2008 and 2018. Patients were treated at the Department of General, Visceral and Transplant Surgery in Mainz, Germany. Additional details have been previously published. 23 This was used as an external test set to determine the final performance metrics. Retrospective use of the patients’ data and material for research purposes was approved by the ethical committee of the medical association of the State of Rhineland-Palatinate (Ref. No. 9888), and informed consent was obtained from all patients included in this study. Patients’ diagnosis and treatment were in accordance with the relevant guidelines in place at the time, and all experiments were in accordance with the Declaration of Helsinki and its later versions. We also followed the REporting recommendations for tumour MARKer prognostic studies (REMARK) guidelines. 24
Preprocessing pipeline
Conventional histopathological glass slides stained with hematoxylin and eosin (H&E) from routine pathology were gathered. Slides from the Mainz cohort were digitized using the Nanozoomer 2.0 HT (Hamamatsu, Japan). Slides from the TCGA cohort were scanned at the respective institution with a variety of different whole slide image (WSI) scanners. After digitalization, slides were annotated at high magnification by placing polygonal regions of interest around the tumorous lesion. This was done under the supervision of board-certified pathologists with special expertise in thyroid/endocrine pathology. Twenty-one of 171 slides of the Mainz cohort and 12 of 496 of the TCGA cohort had to be excluded due to poor quality. Image annotations were used to automatically generate smaller image tiles (1024 × 1024 px) for further preprocessing. Image tiles were normalized to a randomly selected reference image from a case not associated with any of the cohorts using the Reinhard method. 11 Different image augmentations such as mirroring, flipping, limited color distortions, and progressive sprinkles were randomly applied to the training images. Test and validation images were not augmented. When training the model, up to 300 random image tiles per WSI were used to (i) additionally account for class imbalances and (ii) keep computing times within a reasonable time frame. Figure 1 shows a model overview as well as a participant flow diagram.
Molecular characterization
Comprehensive molecular profiling was carried out for both the TCGA and the Mainz cohort. Results of the analyses within the TCGA consortium have previously been described and were gathered via https://portal.gdc.cancer.gov/ and https://www.cbioportal.org/. A detailed description of the underlying Mainz cohort can be found in the study by Staubitz et al. 23 To determine clinically relevant genomic alterations, a multistep pipeline was established. In short, BRAF mutations were determined using polymerase chain reaction (PCR). BRAF and panRAS alterations are mutually exclusive. 25 All cases with BRAF wild type were analyzed using next-generation sequencing (NGS) to identify panRAS mutations. Additionally, all cases were analyzed using fluorescence in situ hybridization (FISH) and fluorescence microscopy to detect RET fusions. The presence of a RET rearrangement was determined by FISH using the ZytoLight® SPEC RET Dual Colour Break Apart Probe (ZytoVison GmbH, Bremerhaven, Germany), as described by Musholt et al. 26 Aberrant signals in the FISH analysis were confirmed using the Archer FusionPlex solid tumor kit (Archer, Boulder, CO, USA) according to the manufacturer’s instructions. Neurotrophic Tyrosine Receptor Kinase (NTRK) fusions were detected by using the Anti-Pan TRK monoclonal antibody, clone EPR17341 (Abcam, Cambridge, MA, USA) in a dilution of 1:250. Immunohistochemistry staining was carried out using a Dako Omnis autostainer (Agilent Technologies, Santa Clara, CA, USA) according to the manufacturer’s instructions. They were also confirmed using NGS. The respective protocols and procedures were established in our certified pathology lab and extensively validated to be used in routine diagnostics in patients with cancer. Different types of alterations with the same clinical implications (e.g., different fusion partners in RET fusion or different types of BRAF mutations) received the same label (“altered”) in order not to make labeling too granular. A detailed list of the alterations can be found in Supplementary Table S1.
Vision Transformer
We established a deep learning algorithm and performed five-fold cross-validation on the discovery cohort (TCGA cases). The most accurate model of the discovery cohort was then used to predict all cases from the external test cohort (Mainz cases) for the final performance estimation. We chose a ViT 20 as our deep learning architecture. The ViT was initialized with pretrained weights (https://huggingface.co/google/vit-base-patch16-224-in21k) and fine-tuned on the discovery cohort. The model was trained for 18 epochs, and the weights of the epoch with the lowest loss were chosen. A ViT is a deep learning model that uses self-attention to weigh the importance of each part of the input. ViTs operate on sequences of data. To turn an image into such a sequence, it is split into fixed-size square patches, each of which is then linearly projected to a vector called a token. A position embedding is added to each token, and a learnable classification token is prepended to the sequence. This sequence is then fed through a transformer encoder. The encoder consists of several blocks of multiheaded self-attention, each followed by a block of fully connected layers with nonlinearities. 20 The output classification token is then passed through a multilayer perceptron head for the classification of the input image. Probabilities for each class were calculated by using softmax on the raw network output. The final classification for each patient was calculated via the weighted average of all their image tiles’ classifications. Markup maps were generated by color coding each tile’s classification and probability to the tile’s x and y position on the whole slide image. Additionally, the highest probability tiles for each class were identified.
Morphological feature analysis and reader study
Morphological features were evaluated by three independent raters, which were blinded to the genomic status of the samples. The following morphological criteria were assessed in the high probability tiles of fusion-associated PTC with fixed variables in a categorical manner by each rater: the intensity of staining of cytoplasm (variables: clear/light or medium/dark), the intensity of staining of nuclei (variables: less than 25% show clear appearance or more than 25% show clear appearance), the presence of calcifications (variables: present or not present), the average size of follicles (variables: no follicles, microfollicular or macrofollicular), and presence of stroma in the tumor (variables: no stroma, less than 30% and more than 30% stroma). After feature identification, a reader study was carried out to determine whether other pathology experts could be educated on the identified histomorphological characteristics to better determine molecular alterations from H&E slides of PTC. In the first round, four raters were provided with 11 whole slide images to classify as either wild type or altered for BRAF, panRAS, or gene fusions. Before proceeding to the second round, the pathologists underwent training in the morphological criteria associated with each alteration. This included an example image for each feature, which was found to be significantly associated with fusion-associated PTC. The second round was then conducted in a similar way but with the added AI-based knowledge.
Statistical analysis
The following metrics were used to evaluate the models: sensitivity, specificity, precision, area under the curve of the precision recall curve (AUPRC), the receiver operating characteristic (AUROC), and the F1 Score defined as
Results
An overview of the model, participant flow diagrams, and the distribution of PTC subtypes can be found in Figure 1. First, we investigated the possibility to predict BRAF mutations from conventional histopathology. It is known that BRAF alterations can be associated with papillary growth pattern and well-developed nuclear features, whereas certain subgroups of follicular variant PTCs can be associated with other alterations. 6,29,30 Upon five-fold cross-validation, the best model achieved an accuracy of 75.3% (66.5–83.5%) (Fig. 2A), an AUROC of 0.834 (0.758–0.905), and an AUPRC 0.814 (0.732–0.887) (Fig. 2B). Cross tables can be found in Figure 2A. On the test cohort, the same model achieved an accuracy of 79.3% (72.7–85.5%), an AUROC of 0.882 (0.829–0.931), and an AUPRC 0.896 (0.847–0.940) (Fig. 2B). Cross tables can be found in Figure 2A. BRAF alterations could easily be visualized within the H&E whole slide image by using our established classification markups (Fig. 2C). Next, we looked at panRAS mutations as it is known that up to 15% of BRAF wild type can be panRAS mutated, 31 whereas BRAF and panRAS alterations are mutually exclusive. 25 Correspondingly, panRAS alterations show a follicular growth pattern and fewer classical nuclear features compared with BRAF mutations. 31 Upon five-fold cross-validation, the best model achieved an accuracy of 88.7% (82.2–94.7%), an AUROC of 0.924 (0.870–0.975), and an AUPRC 0.432 (0.330–0.536) (Fig. 2F). Cross tables can be found in Figure 2E. On the test cohort, the same model achieved an accuracy of 89.3% (84.2–94.0%), an AUROC of 0.876 (0.822–0.927), and an AUPRC 0.332 (0.260–0.413) (Fig. 2F). Cross tables can be found in Figure 2E. Again, panRAS alterations could easily be visualized within the H&E whole slide image by using our established classification maps/markups (Fig. 2G). High probability tiles for both genes showed a classical inverse pattern where BRAF mutated and panRAS wildtype cases would display papillary morphology, while BRAF wild type and panRAS mutated cases would display follicular growth (Fig. 2D, H). Of note, we did not distinguish between different types of BRAF or panRAS mutations (e.g., RAS-like BRAF mutations), which could explain some false predictions (Supplementary Fig. S1A).
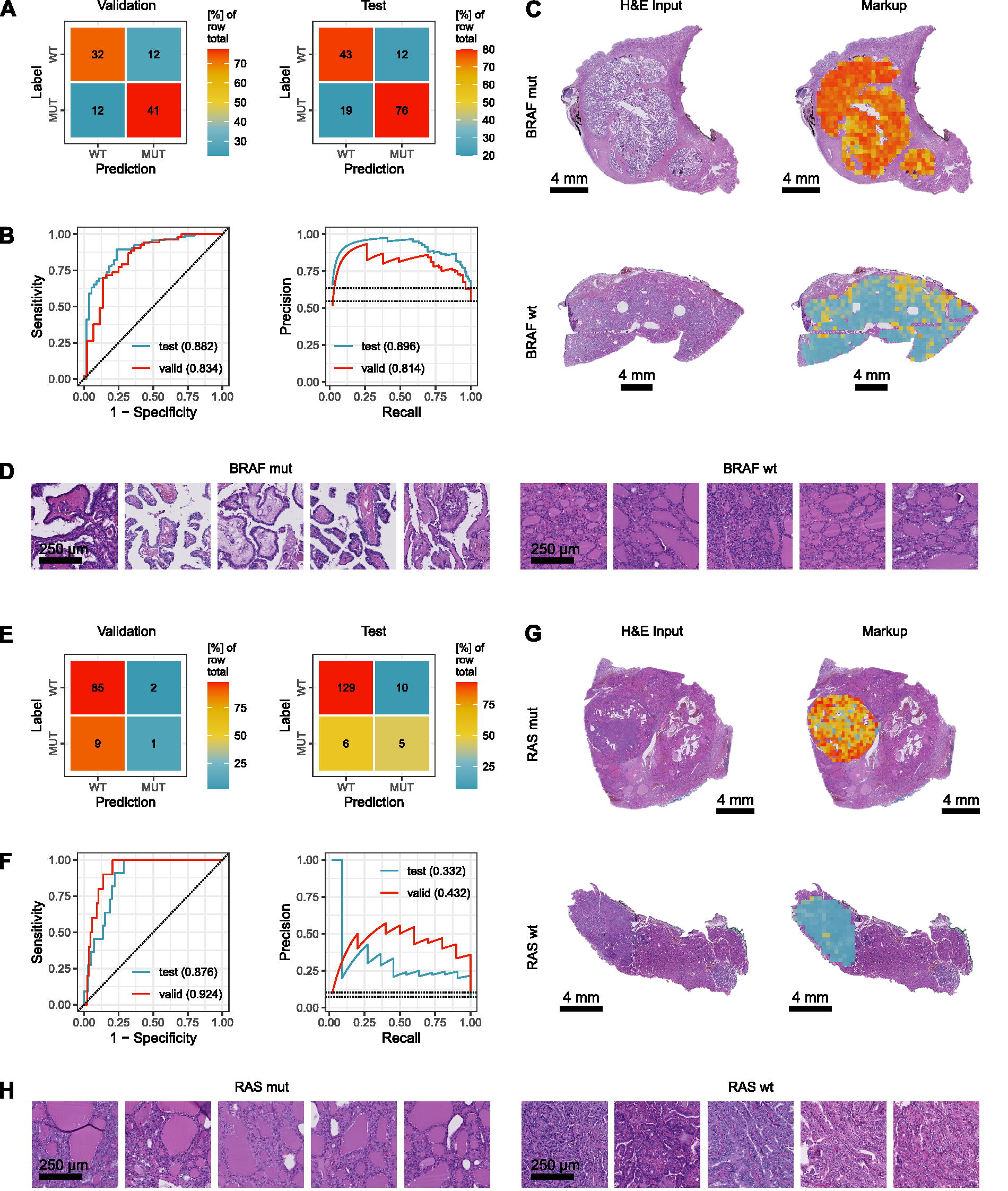
Prediction of BRAF
In the next step, we investigated the possibility to predict fusion-associated PTC. As previously described, three different types of RET alterations could be identified in the Mainz cohort, namely RET fusions with CCDC6, NCOA4, and ERC1 as fusion partners. Additionally, one TPM3-NTRK1 fusion was found. These different fusion events were collectively classified as one class. Upon five-fold cross-validation, the best model achieved an accuracy of 88.7% (82.2–94.7%), an AUROC of 0.876 (0.809–0.939), and an AUPRC 0.487 (0.381–0.588) (Fig. 3B). Cross tables can be found in Figure 3A. On the test cohort, the same model achieved an accuracy of 84.7% (78.8–90.2%), an AUROC of 0.858 (0.801–0.912), and an AUPRC 0.278 (0.213–0.353) (Fig. 3B). Cross tables can be found in Figure 3A. Fusion-associated PTC could easily be visualized within the H&E whole slide image by using our established classification maps/markups (Fig. 3C). This corresponds with areas of genomic alterations as detected by FISH, for example (Supplementary Fig. S1B).
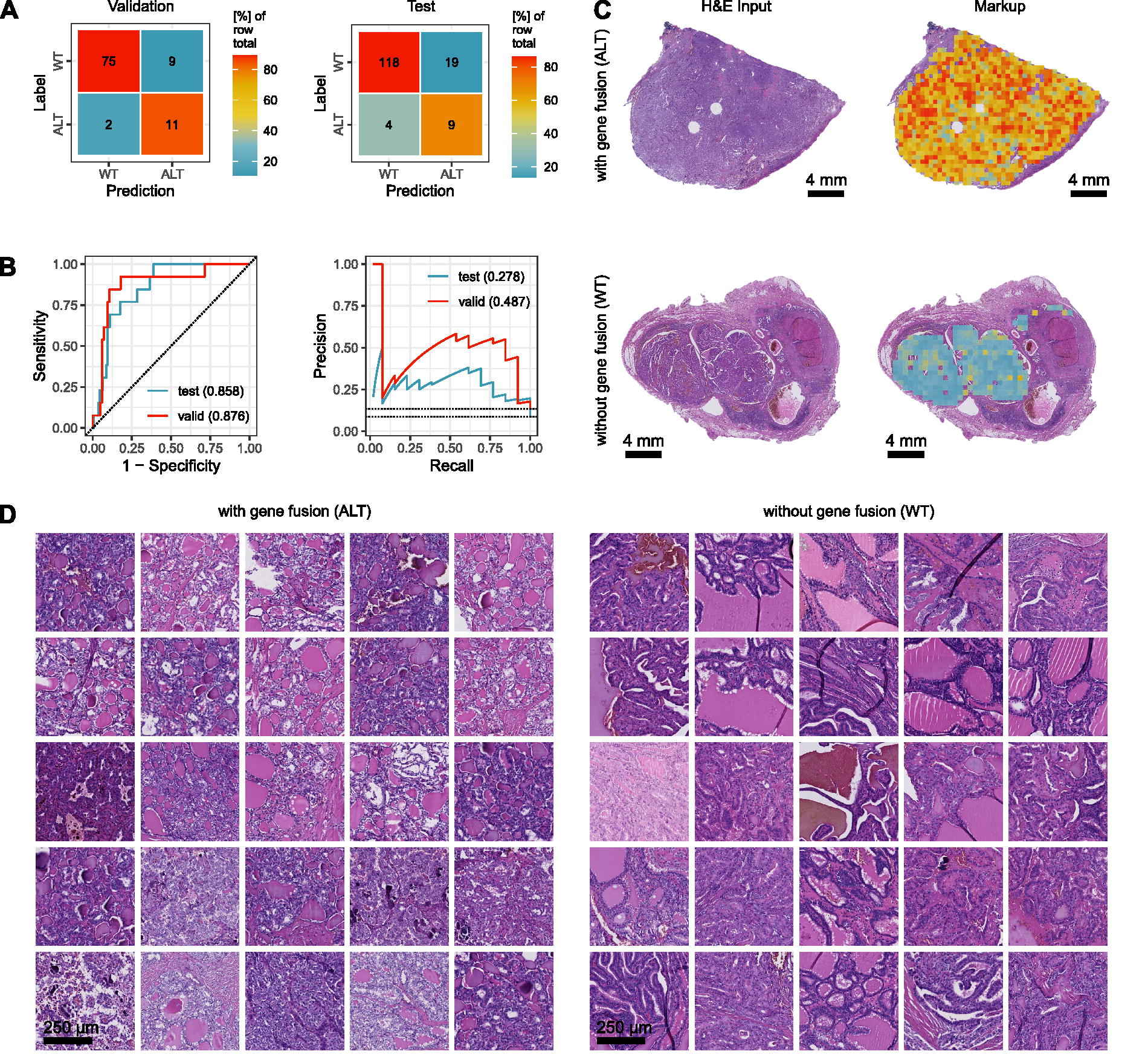
Prediction of fusion-associated PTC.
Next, we investigated whether the deep learning model could help to discover histomorphological features associated with fusion-associated PTC. To this end we first examined the 25 image tiles of the Mainz cohort with the highest and the lowest probability for fusion events. This led to the identification of new, previously undescribed features of wild-type and altered tumors. The presence of calcifications, clear cytoplasm, and small sized follicles was associated with fusions (Fig. 3D). Vice versa, tumors with larger follicles and darker cytoplasm were associated with wild type (Fig. 3D, 4B). To confirm this descriptive finding, three pathology experts were tasked with scoring these and other criteria within the test cohort in a blinded fashion. Here, we found a significant association of these features with fusion status (Fig. 4A, B). Similarly, top-level tiles confirmed known features for BRAF and panRAS mutated PTC (Fig. 2D, H). After having identified novel features associated with fusion-associated PTC using ViTs, we intended to find out whether pathology experts could be educated on these features to better identify molecular alterations from histopathological slides. Interestingly, participants of the reader study improved their accuracy significantly from around 50% (similar to random guessing) to over 80%. This effect can also be seen in the ROC and PRC curves (Fig. 4C, D). For BRAF and panRAS alterations with already known morphological features, this effect could not be observed (Supplementary Fig. S2).
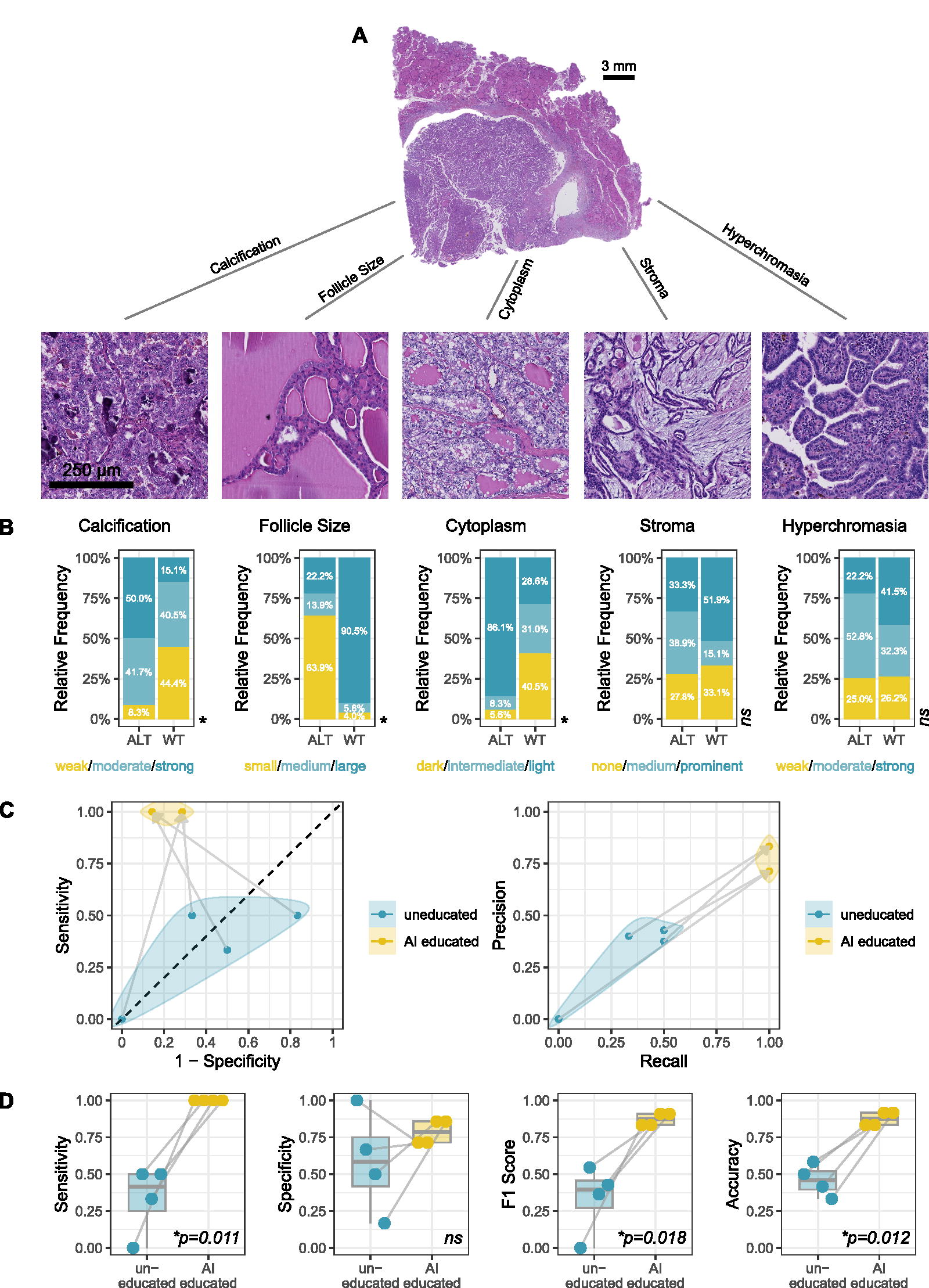
PTC tissue properties and education study.
Discussion
Extensive genetic testing of tumor tissues is costly and complex and poses significant demands on health care systems. Nonetheless, it would be ethically problematic to deny patients access to the most advanced diagnostic and therapeutic technologies. This is particularly true for thyroid cancer, which often affects younger patients and where molecular alterations have important implications on diagnosis, prognosis, and treatment. AI and machine learning can offer crucial support to pathologists and have the potential of making the diagnostic process more cost-effective and thereby improving patient care, even in economically disadvantaged regions. The results of our study are significant in the following regards: (i) They confirm previous data on predicting BRAF and RAS alterations from conventional histopathology, 32 this time using a state-of-the-art AI algorithm and a true, well characterized, independent, external test set. This is absolutely necessary for future clinical application. 33 In this study we further provide a complete performance assessment, showing that our model’s predictions are balanced and not falling back on just predicting the majority class of an imbalanced dataset. (ii) For the first time, we included other additional clinically relevant genetic alterations, such as gene fusions. This broadens the spectrum of potential clinical applications as gene fusions can be targeted by specific treatments—for example., with selpercatinib (RET) or larotrectinib (NTRK). (iii) With the help of various explainability approaches, we not only confirmed morphological criteria that are known to be associated with BRAF and panRAS mutations. 29 We also identified new histopathological features that are associated with gene fusions in PTC. There are few recent studies investigating the morphology of PTCs harboring RET alterations, for example; however, no clear picture could be drawn from these. 34,35 Although it’s important for an AI’s decision-making processes to be comprehensible, on the contrary, some argue that a well-performing AI tool doesn’t have to be completely explainable to be useful. 36,37 Our research demonstrates that explainability approaches can provide valuable insights into the process of mutational analysis. Interestingly, the newly discovered features associated with fusions in PTC are reminiscent of the morphology of other translocation-associated tumors (e.g., TFE3-associated renal cell carcinomas). 38 This could point to a common mechanism associated with this phenotype. Wollek et al. 39 were able to show that attention-based heat maps of ViTs are structurally easier for physicians to interpret compared with convolutional neural networks (CNNs).
There are important limitations to our study. Human annotations can be time-consuming and might introduce potential bias. However, we and others argue that thorough annotation can boost accuracy especially in light of limited case numbers. 40 Future studies could use a two-step approach by first identifying tumor tissue and then making the molecular classification. Additionally, weakly supervised methods could also be explored. 41 It’s also worth noting that previous research of Tsou et al. 19 has shown a somewhat better performance in the classification of some alterations. However, it remains to be seen if this would hold true upon a more extensive external validation/testing. Other crucial limitations of our study are that we were not able to show any prognostic capabilities of our models and that it was performed in a retrospective manner. Furthermore, we did neither include any additional information about different PTC subtypes, nor did we distinguish between different types of mutations (BRAF-like, RAS-like, etc.) or fusions. Future efforts should therefore also focus on training additional, more refined AI models for these types of predictions, which could then potentially be used for prognostication in PTC.
In conclusion, our study demonstrates that clinically relevant genomic alterations can be predicted using conventional histopathology and state-of-the-art AI. Together, these alterations account for approximately 70% of the actionable genomic changes of patients with PTC. Furthermore, comprehensive analysis of AI-based morphological features has confirmed known histopathological patterns associated with certain molecular changes as well as discovered completely new ones. While there is a strong need for prospective research, if confirmed, this could potentially help pathologists to suspect certain alterations earlier during daily routine and better guide molecular diagnostics.
Declaration of Generative AI and AI-Assisted Technologies in the Writing Process
During the preparation of this work, the author(s) used DeepL and ChatGPT for checking language and grammar. After using this tool/service, the author(s) reviewed and edited the content as needed and take(s) full responsibility for the content of the publication.
Footnotes
Authors’ Contributions
I.M.: Conceptualization, validation, analysis, investigation, data curation, and writing (original draft). S.S.: Conceptualization, methodology, software, validation, analysis, investigation, and data curation. S.F.: Conceptualization, methodology, software, validation, analysis, investigation, writing (original draft), visualization, supervision, and project administration. C.G.: Methodology, software, and visualization. M.E.: Investigation and writing (review and editing). A.F.: Investigation. M.O.M.: Investigation. S.M.: Investigation and writing (review and editing). M.M.G.: Investigation and writing (review and editing). S.S.: Investigation. D.-C.W. Investigation. A.S.: Investigation and resources. M.J.: Investigation and writing (review and editing). C.M.: Resources and data curation. N.H.: Resources and data curation. T.J.M.: Resources, data curation, and writing (review and editing). J.I.S.-V.: Resources and data curation. J.N.K.: Writing (review and editing). D.T.: Writing (review and editing). W.R.: Writing (review and editing) and supervision. All authors approved the final version of the article to be published and are accountable for this work.
Disclaimer
The views and opinions expressed are those of the author(s) only and do not necessarily reflect those of the European Union. Neither the European Union nor the granting authority can be held responsible for them.
Author Disclosure Statement
J.N.K. declares consulting services for Owkin, France; DoMore Diagnostics, Norway; Panakeia, UK; Scailyte, Switzerland; Cancilico, Germany; Mindpeak, Germany; and Histofy, UK; furthermore, he holds shares in StratifAI GmbH, Germany, and has received honoraria for lectures by AstraZeneca, Bayer, Eisai, Janssen, MSD, BMS, Roche, Pfizer, and Fresenius. D.T. received honoraria for lectures by Bayer and holds shares in StratifAI GmbH, Germany. M.E. declares personal fees, travel costs, and speaker’s honoraria from MSD, AstraZeneca, Janssen-Cilag, Cepheid, Roche, Astellas, and Diaceutics; research funding from AstraZeneca, Janssen-Cilag, STRATIFYER, Cepheid, Roche, Gilead, and Owkin; and advisory roles for Diaceutics, MSD, AstraZeneca, Janssen-Cilag, GenomicHealth, Owkin, and Gilead. S.F. has received honoraria from MSD and BMS. The other authors declare no conflicts of interest.
Funding Information
J.N.K. is supported by the German Federal Ministry of Health (DEEP LIVER, ZMVI1-2520DAT111), the German Cancer Aid (DECADE, 70115166), the German Federal Ministry of Education and Research (PEARL, 01KD2104C; CAMINO, 01EO2101; SWAG, 01KD2215A; TRANSFORM LIVER, 031L0312A; TANGERINE, 01KT2302 through ERA-NET Transcan), the German Academic Exchange Service (SECAI, 57616814), the German Federal Joint Committee (TransplantKI, 01VSF21048), the European Union’s Horizon Europe and innovation program (ODELIA, 101057091; GENIAL, 101096312), and the National Institute for Health and Care Research (NIHR, NIHR213331) Leeds Biomedical Research Centre. S.F. is supported by the German Federal Ministry of Education and Research (SWAG, 01KD2215C), the German Cancer Aid (DECADE, 70115166 and TargHet, 70115995), and the German Research Foundation (504101714). M.J. is supported by the German Cancer Aid (TargHet, 70115995). M.E. is supported by the Else Kröner-Fresenius-Stiftung/EKFS (2020_EKEA.129; 2023_EKES.07), the German Federal Ministry of Education and Research (HANCOCK, 01KD2211B), the IZKF of the FAU Erlangen-Nürnberg (Clinician Scientist Program; TOPeCS T04; advanced grant IZKF-D41), and the Bavarian Cancer Research Center/BZKF (YSF-TP01; INITIATOR BF/01/E/Pila). M.M.G. is supported by the German Research Foundation (Project Number 318346496, SFB1292/2 TPQ1 and TP22). This work was funded by the European Union. The other authors declare no funding.
Supplementary Material
Supplementary Data
Supplementary Figure S1
Supplementary Figure S2
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.