
Abstract
Background:
Thyroid eye disease (TED) is a sight-threatening autoimmune disease with cigarette smoking as one of the key risk factors. Cigarette smoking affects both the severity of TED and the patient’s response to medication. However, the underlying pathogenic mechanisms of smoking in TED remain unclear.
Methods:
Orbital fibroblasts (OFs) were extracted from patients with TED and non-TED controls, and treated with cigarette smoking extract (CSE). Luminex assays and Western blots were employed to examine inflammatory status and pathological phenotypes of OFs. A specific reactive oxygen species (ROS) probe was used to evaluate oxidative stress levels. RNA-sequencing of CSE-treated OFs was used to analyze differentially expressed genes. Immunofluorescence and RNA-sequencing were used to examine the expression of receptor for advanced glycation end products (RAGE) signaling molecules in patients. Small interfering RNA sequences and a RAGE-specific inhibitor were employed to investigate the effects of RAGE blockade on cigarette smoking-related pathological phenotypes. To validate our findings in vivo, we generated an adenovirus-induced TED mouse model with exposure to cigarette smoke.
Results:
Exposure to CSE resulted in an inflammatory phenotype of OFs together with higher levels of oxidative stress. OFs exposed to CSE presented susceptibility to transforming growth factor-β-induced myofibroblast differentiation, and 15-D-PGJ2-induced adipocyte differentiation, indicating pro-fibrotic and pro-adipogenic phenotypes. RNA-sequencing of CSE-treated OFs revealed upregulation of RAGE signaling molecules. TED patients with smoking history also exhibited higher levels of RAGE signaling, both in the orbit and peripheral blood, compared with non-smoking patients. Enhancement of inflammatory status was associated with activation of the ROS-nuclear factor-kappa B pathway downstream of RAGE. RAGE gene interference or administration of RAGE inhibitor effectively mitigated cigarette smoking-related pathological changes in OFs. Disrupting RAGE signaling in TED mice efficiently ameliorated smoking-induced disease progression in vivo.
Conclusions:
Cigarette smoking-relevant TED progression was linked with RAGE signaling activation, leading to the exacerbation of orbital inflammation and tissue-remodeling, including fibrosis and adipogenesis. Our findings demonstrate that cigarette smoke exposure affects the biological characteristics of TED-derived OFs and supports RAGE as a promising therapeutic target for the management of patients with TED and smoking habits.
Introduction
Thyroid eye disease (TED), also known as Graves’ orbitopathy, is an organ-specific autoimmune condition usually related to thyroid dysfunction, particularly hyperthyroidism of Graves’ disease 1 –3 ; however, its pathogenesis is rather complex. Studies suggest that orbital fibroblasts (OFs) expressing the autoantigen thyrotropin (TSH) receptor (TSHR) may contribute to TED pathology. 1,2 Upon stimulation with autoantibodies, OFs produce various inflammatory cytokines and hyaluronan (HA), leading to orbital inflammation and extracellular matrix deposition. 3,4 OFs can also differentiate into either myofibroblasts or adipocytes, causing orbital fibrosis or adipogenesis. 4,5 Different T-cell subtypes, such as T helper (Th)1, Th2, and Th17, are also implicated in the inflammation and pathological changes of TED. 6
As an autoimmune disease, TED is influenced by many environmental factors. 7 Cigarette smoking is a modifiable risk factor that contributes to disease development. 8 Smoking greatly increases the incidence of TED and is related to more severe eye signs. 8,9 International guidelines and consensuses agree that giving up smoking is essential for TED management. 7,10 However, the mechanisms underlying cigarette smoking in TED progression are yet to be examined. A study in smoking patients with TED found elevated oxidative stress levels, 11 while an in vitro study showed upregulation of heme oxygenase-1 in OFs treated with cigarette smoke extract (CSE), indicating a higher level of reactive oxygen species (ROS). 12 Furthermore, CSE-treated OFs expressed fibronectin and CCAAT-enhancer-binding proteins α and β, suggesting potential pro-fibrotic and pro-adipogenic effects of smoking. 12,13
In this study, we aimed to investigate the pathological effects and underlying mechanisms of cigarette smoke on TED, with an emphasis on the activation of receptor for advanced glycation end products (RAGE) in OFs. A deeper understanding of the molecular alterations in smoking-related pathology may provide valuable insights into potential therapeutic strategies for patients with TED and smoking habits.
Materials and Methods
CSE preparation
CSE was collected by bubbling the smoke from two filtered, commercially available Chienmen-brand cigarettes into 50 mL serum-free Dulbecco’s modified Eagle’s medium (DMEM, Gibco) using a mechanical vacuum pump, and was sterilized by filtering through a 0.22-μm filter (Millipore). Such concentration was set as 1 CSE in the following study. Quantity of cigarettes and the negative pressure suction flow rate were maintained consistent among baches. The CSE was stored at 4°C at the same pH as untreated DMEM.
OF extraction
Orbital tissues were cut into small pieces and digested with collagenase I (1 mg/mL) (Sigma) at 37°C immediately after collection. OFs from these tissues were seeded into six-well plates and cultured in DMEM containing 10% fetal bovine serum (Gibco), 100 IU/mL penicillin, and 100 mg/mL streptomycin (Gibco) at 37°C in a 5% CO2 humidified incubator. All the in vitro experiments in the study were conducted with three OF strains derived from three independent donors (demographic characteristics summarized in Supplementary Table S1).
TED mouse model
Six-week-old female BALB/c mice were obtained from Shanghai Model Organisms Center, Inc. Animals were housed in a vivarium on a 12-h light-dark cycle at 21°C and 55% humidity. The TED model was established following the protocol in a previous study. 14
The mice were randomly divided into four groups: 10 were injected with adenovirus expressing human TSHR A subunit (Ad-hTSHRA); 10 received Ad-hTSHRA and were exposed to 1 h of smoke from five burning cigarettes in a smoking chamber per day; 10 received Ad-hTSHRA, 1 h of daily CSE exposure as above, and an intraperitoneal injection of FPS-ZM1 (1 μg/g of bodyweight) (Selleck) once a week; and 10 controls received the same dose of adenovirus expressing enhanced green fluorescent protein (Ad-EGFP). After nine injections, an equal number of mice were randomly selected from each group for further evaluation under blinded manners by independent operators.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 9.5.1 software. Comparisons were assessed using Student’s t test or one-way analysis of variance, as appropriate. Quantified data are presented as means ± standard deviation of biological triplicates. A p-value <0.05 was considered statistically significant.
A detailed experimental scheme is provided in Supplementary Data.
Results
CSE induces oxidative stress and inflammatory phenotypes in OFs
To uncover the impacts of smoking on TED-derived OFs, we treated the cells with CSE-conditioned culture medium, in accordance with previous reports. 13,15 Our initial evaluation of cytotoxicity using Cell Counting Kit-8 assays showed little effect of CSE on OFs viability (Supplementary Fig. S1A). Disordered HA accumulation is a cardinal feature of TED. 16 Using Enzyme linked immunosorbent assay (ELISA), we demonstrated CSE concentration-dependent HA expression in the supernatants of OFs, indicating that CSE induced the secretion of extracellular matrix materials and regional edema (Supplementary Fig. S1B). Based on cell viability results, we applied 1/4 CSE concentration in subsequent experiments to ensure minimal impact on cell viability. Next, we examined the cytokine/chemokine profiles in OF supernatants. Compared with unstimulated cells, a 48-h CSE treatment significantly enhanced the secretion of various cytokines and chemokines, namely interleukin (IL)-6, IL-8, tumor necrosis factor (TNF)-α, vascular endothelial growth factor, monocyte chemoattractant protein 1, chemokine (C-C motif) ligand (CCL)9, CCL20, and granulocyte-macrophage colony-stimulating factor (Fig. 1A and Supplementary Fig. S1C). A 24-h CSE treatment significantly increased IL-6, TNF-α, and CCL20 (Fig. 1A), which reportedly play vital roles in orbital inflammation. 17 Given that smoking results in high levels of oxidative stress, 11 we performed an oxidative stress evaluation of OFs from patients with TED and non-TED controls with a fluorescent ROS probe, which revealed that CSE induced elevated ROS levels at 24 h (Fig. 1B and C). Such effects were more pronounced in TED OFs compared with non-TED OFs. Taken together, these results confirmed that exposure to CSE initiates inflammatory phenotypes accompanied by an aggravated oxidative stress status.
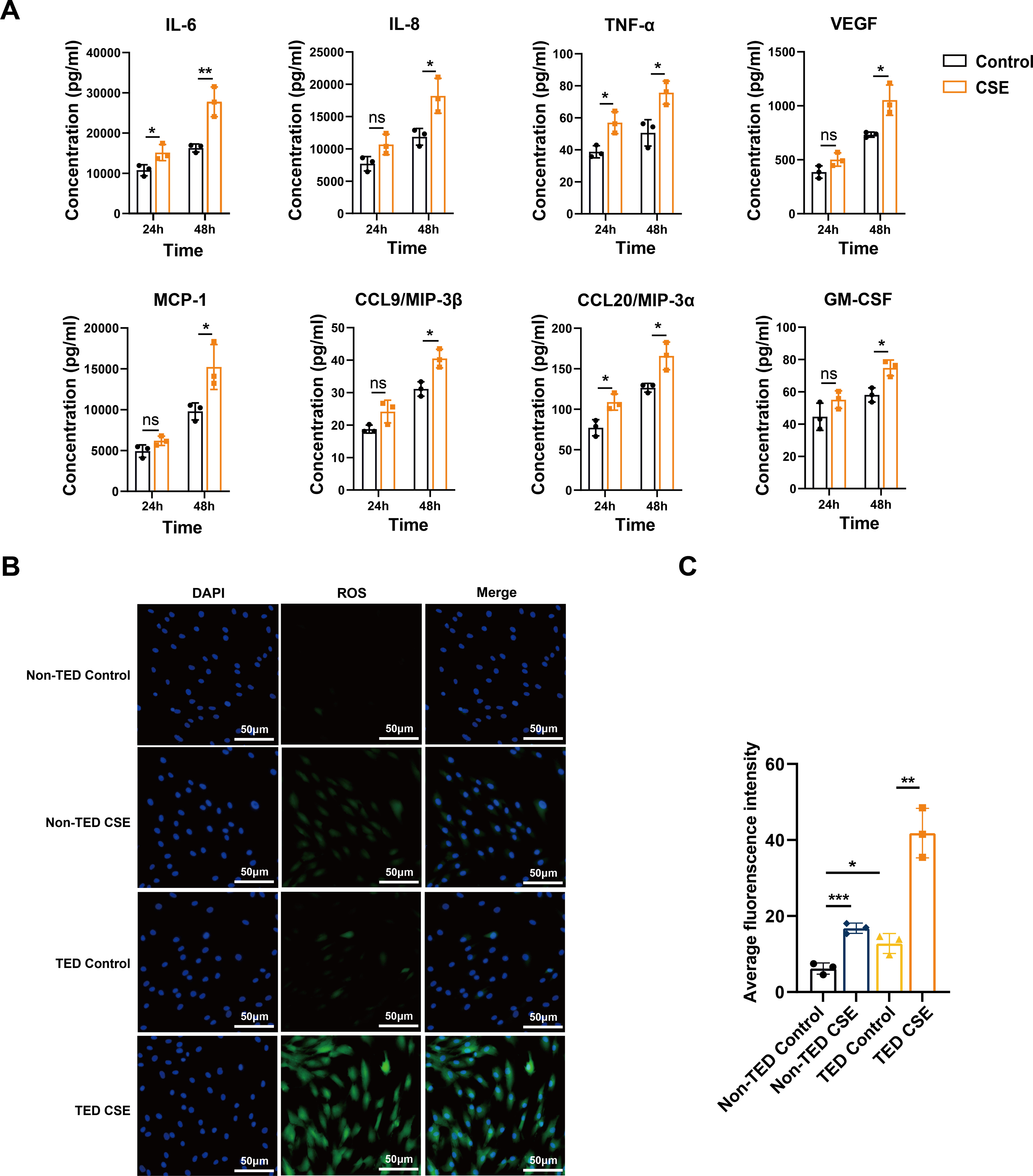
CSE leads to upregulation of ROS and induces inflammatory phenotypes in OFs.
CSE exacerbates fibrotic and adipogenic differentiation of OFs
To evaluate the effects of CSE on fibrotic differentiation, we employed recombinant human transforming growth factor (rhTGF)-β to induce fibrosis in TED OFs, as previously reported. 5,18 Fibrotic markers including fibronectin, collagen I, and tissue inhibitor of metalloproteinase 1 (TIMP-I) 19,20 were detected using Western blots. We observed that CSE promoted fibrotic phenotypes of OFs, even in the absence of TGF-β stimulation (Fig. 2A and B), implying a pro-fibrogenic effect of cigarette smoke itself. The combination of CSE and TGF-β promoted the expression of fibrotic markers more strongly than TGF-β (Fig. 2A and B). To further investigate the role of CSE in TED adipogenesis, we used 15-D-PGJ2, as previously reported. 5 OFs showed differentiation into adipocytes marked by the activation of peroxisome proliferator-activated receptor (PPAR)-γ (Fig. 2C and D). CSE did not directly promote adipogenic differentiation of OFs, but did significantly enhance 15-D-PGJ2-induced PPAR-γ activation, as well as perilipin A and adiponectin expression (Fig. 2C and D). Oil red O staining further showed that CSE plus 15-D-PGJ2 drastically enhanced lipid droplet formation (Fig. 2E and F). These results implied that CSE contributes to TED tissue-remodeling via enhanced fibrosis and adipogenesis in OFs.

CSE induces exacerbated pathological changes in OFs.
CSE induces upregulation of RAGE signaling in TED-derived OFs
RNA-sequencing of TED OFs at different timepoints of CSE treatment (6, 8, 12, 24, 48, and 72 h) revealed a continuous increase in the number of differentially expressed genes (Fig. 3A). Kyoto Encyclopedia of Genes and Genomes analysis showed enrichment of the advanced glycation end products (AGE)-RAGE signaling pathway at 12, 24, 48, and 72 h in the CSE treatment groups (Fig. 3B and Supplementary Fig. S2A). Next, we profiled key genes downstream of RAGE signaling. Inflammation-related genes (IL6, CXCL8, TNF, and IL1B), fibrosis-related genes (TGFBR1, TGFB2, TGFB3, COL1A1, and COL3A1), and the oxidative stress-related gene HMOX1 were upregulated at different timepoints of CSE treatment (Fig. 3C). Additionally, gene set enrichment analysis revealed that genes involved in lipid metabolism (adipogenesis, fatty acid metabolism, and cholesterol homeostasis) were enriched in the CSE treatment groups (Supplementary Fig. S2B). These results supported the idea that RAGE activation contributes to smoking-related pathological phenotypes. To verify this hypothesis, we performed immunofluorescence for RAGE expression in orbital tissues collected from age- and sex-matched patients with TED with or without smoking habits, and non-TED controls (demographic characteristics summarized in Supplementary Table S2). Smoking patients with TED exhibited higher levels of RAGE fluorescence signals compared with non-smoking patients and non-TED controls (Fig. 3D and E). Moreover, correlation analysis suggested a positive association between orbital RAGE fluorescence intensity and the patients’ smoking index (Supplementary Fig. S2C). Likewise, the transcriptomes of peripheral blood mononuclear cells (PBMCs) from patients with TED displayed enrichment of the AGE-RAGE pathway in the smoking group (Fig. 3F; demographic characteristics summarized in Supplementary Table S3). Additionally, TED-derived OFs with CSE stimulation showed higher levels of RAGE induction compared with untreated TED-derived OFs and non-TED-derived OFs (Fig. 3G and H). We also noticed the baseline expression levels of RAGE were higher in TED OFs than in non-TED OFs (Fig. 3G and H), suggesting it may serve as an important pathogenic factor.
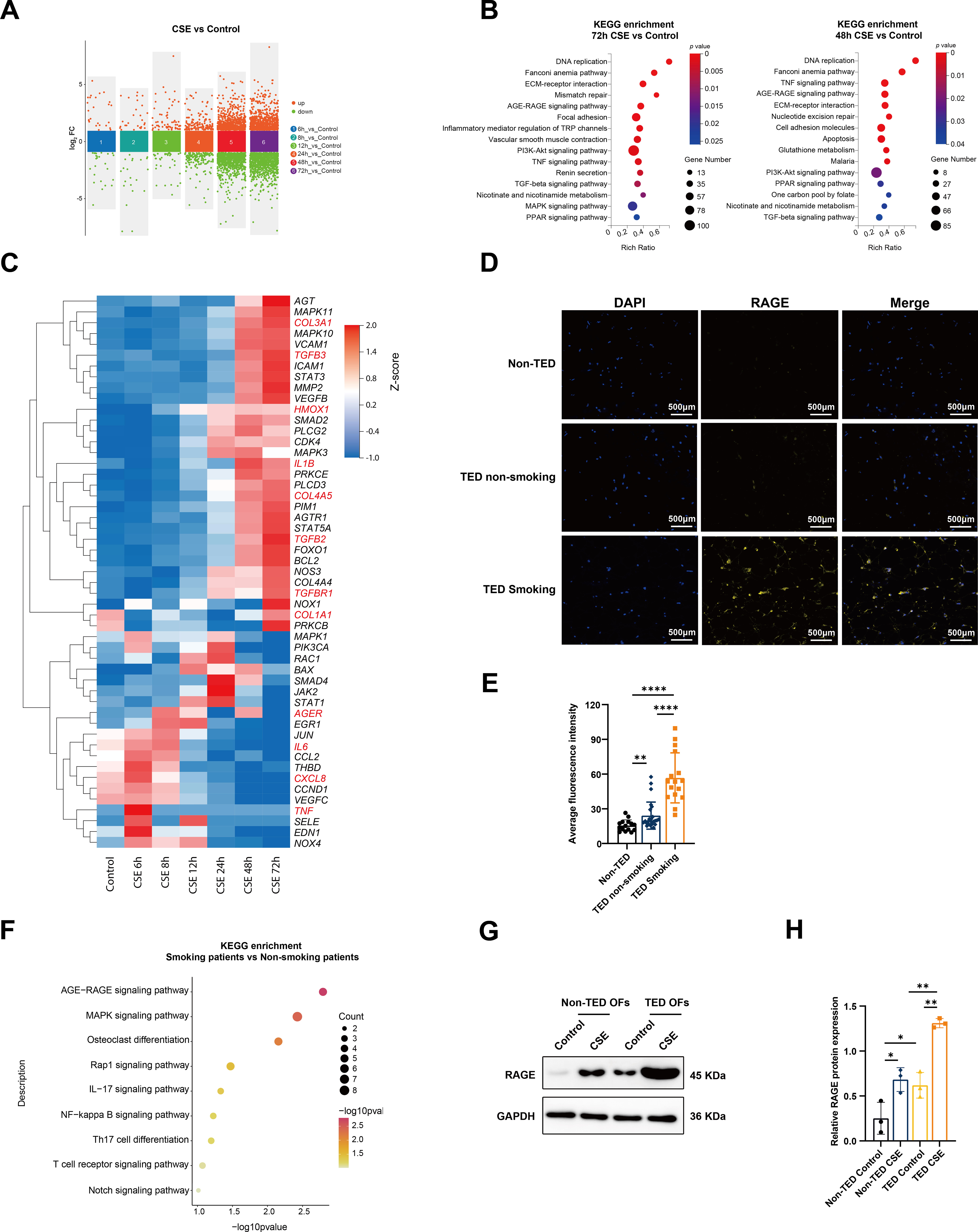
Smoking correlates with RAGE signal upregulation in TED.
Disruption of RAGE signaling effectively alleviates CSE-induced oxidative stress and inflammatory responses
We synthesized two small interfering (siRNA) sequences targeting RAGE (Supplementary Fig. S3A and B) and employed these siRNAs, along with the RAGE-specific inhibitor FPS-ZM1, 21 to determine if RAGE blockade could downregulate smoking-induced changes. Results showed that most of the inflammatory cytokines and chemokines upregulated by CSE were significantly reduced by RAGE inhibition at 48 h (Fig. 4A). Additionally, ROS detection illustrated that RAGE blockade effectively decreased the CSE-mediated upregulation of oxidative stress (Fig. 4B and C). To gain insight into the relationships between RAGE, ROS production, and the inflammatory process, we further analyzed the transcriptome data, revealing enrichment of nuclear factor-kappa B (NF-κB) signaling in OFs stimulated with CSE in early (6 and 8 h) timepoint groups (Fig. 4D and Supplementary Fig. S3C). Previous studies indicated that NF-κB signaling lies downstream of the RAGE receptor, playing a key role in AGE-RAGE-relevant inflammation. 22,23 We also found that CSE significantly increased the phosphorylation of IκB and enhanced translocation of the NK-κB p65 subunit into the nucleus at 8 h (Fig. 4E and F). Such activation of NK-κB signaling by CSE was significantly inhibited by RAGE interruption (Fig. 4E and F). These results suggested that smoke-activated RAGE promoted inflammatory cytokine production in TED-derived OFs through NF-κB signaling.
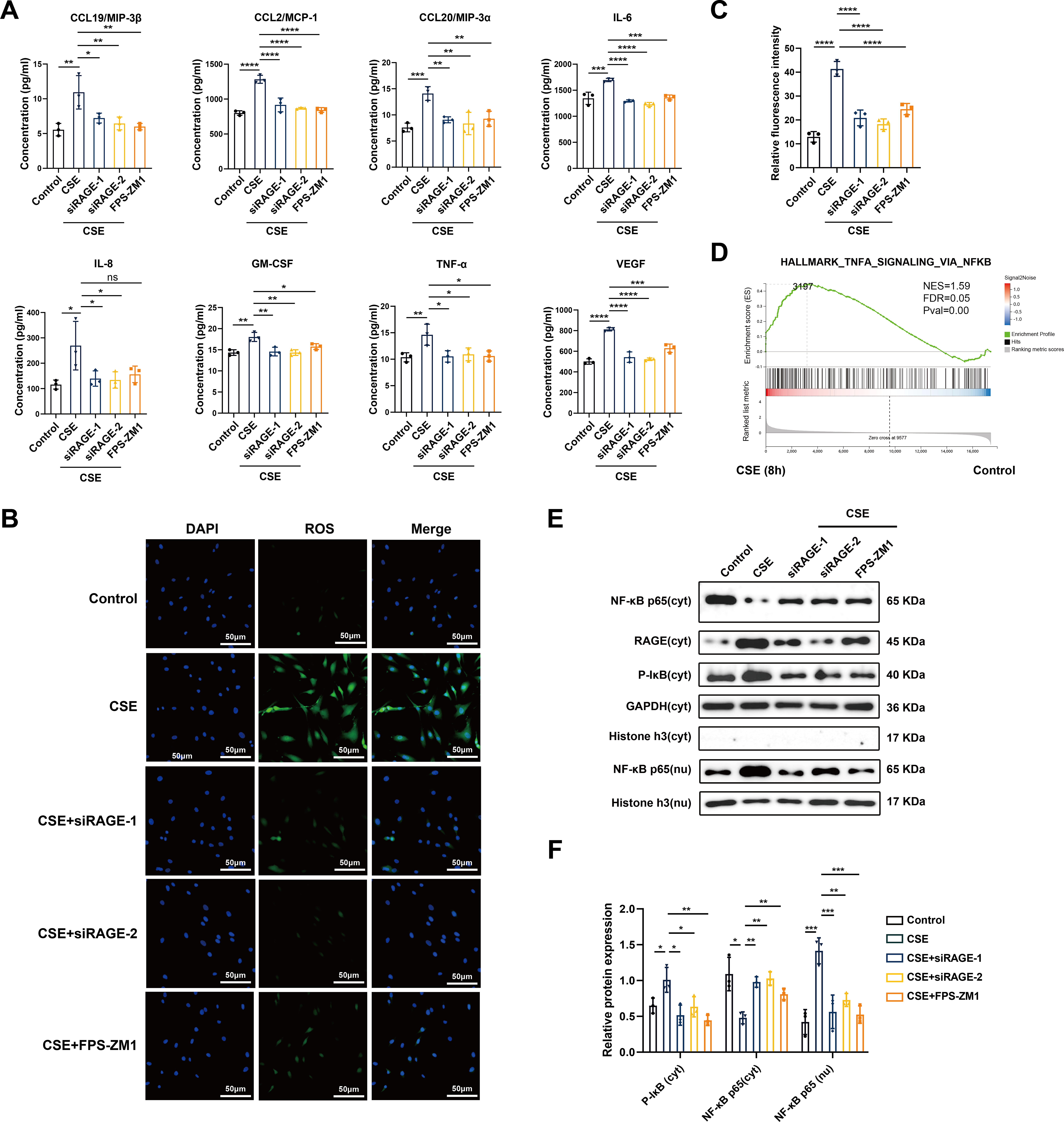
CSE induces inflammatory phenotypes in OFs through activation of RAGE-ROS-NFκB signaling.
Disruption of RAGE signaling remarkably attenuates CSE-exacerbated fibrotic and adipogenic pathological changes
Next, we defined the potential of RAGE blockade for the management of smoking-enhanced fibrosis and adipogenesis in OFs. Our data showed that siRNAs and FPS-ZM1 significantly downregulated the expression levels of fibrotic markers triggered by combination treatment with CSE and rhTGF-β in OFs (Fig. 5A and B). Meanwhile, siRNAs and RAGE inhibition also significantly downregulated the expression levels of adipogenic markers triggered by treatment of OFs with CSE plus 15-D-PGJ2 (Fig. 5C and D). Oil red O staining of OFs further supported the amelioration of CSE-induced adipogenesis by RAGE disruption (Fig. 5E and F). These data indicated that inhibition of RAGE signaling could improve orbital tissue-remodeling caused by cigarette smoking in TED.

Blockade of RAGE efficiently inhibits CSE-induced aggravation of pathological changes in OFs.
RAGE disruption is an effective therapeutic strategy for smoking-induced disease aggravation in TED mice
To confirm our in vitro findings, we generated an autoimmune TED mouse model induced by Ad-hTSHRA, as previously reported. 14,24,25 Figure 6A illustrates the smoking platform commonly used for mouse cigarette smoke exposure, as referenced in respiratory disease studies, which effectively simulates human smoking behavior. 26 –29 Mice were randomly separated into four treatment groups, immunized with Ad-EGFP (control), Ad-hTSHRA alone (TED), or Ad-hTSHRA with smoke exposure, with or without FPS-ZM1 treatment (Fig. 6B). After 34 weeks, TED mice exposed to daily cigarette smoke showed significant bodyweight loss (Fig. 6C). Pulmonary function tests confirmed the long-term inhalation of smoking particles (Supplementary Fig. S4A–C). Exposure to cigarette smoke also exacerbated thyroid function abnormalities in TED mice, including altered levels of thyroid receptor antibodies, total T4 (TT4), and TSH. Treatment with FPS-ZM1 slightly relieved these thyroid function changes, but not significantly (Supplementary Fig. S4D–F). Next, we evaluated the groups for eye signs. The TED group presented classical manifestations, including eyelid inflammation and proptosis, with smoking drastically exacerbating regional inflammation, as indicated by severe inflammatory edema of the eyelid (Fig. 6D). FPS-ZM1 treatment effectively alleviated these eye signs (Fig. 6D). Because severe eyelid edema made it hard to evaluate the extent of proptosis in the smoking group, we performed magnetic resonance imaging (MRI) of intraorbital tissues, which revealed significantly more proptosis in the smoking TED group than in the non-smoking TED group (Fig. 6E and F). This proptosis was effectively mitigated by FPS-ZM1 (Fig. 6E and F). Furthermore, the increased thickness of mouse extraocular muscles (EOM) observed in smoking TED mice was dampened by FPS-ZM1 (Supplementary Fig. S4G and H). Eyelid inflammation, as indicated by eyelid thickness, was also mitigated in the FPS-ZM1-treated smoking TED group (Supplementary Fig. S4I and J). Moreover, hematoxylin and eosin staining of mouse orbital tissues showed that smoking significantly enhanced orbital fat proliferation in TED mice, but this fat expansion was dampened by FPS-ZM1 treatment (Fig. 6G and H). EOM fibrosis was examined with Masson staining, which confirmed the therapeutic effects of the RAGE inhibitor (Fig. 6I and J). Collectively, our findings with this preclinical model of TED mice exposed to smoking demonstrated the potential of RAGE as a target for the management of smoking-related damage to the orbit.
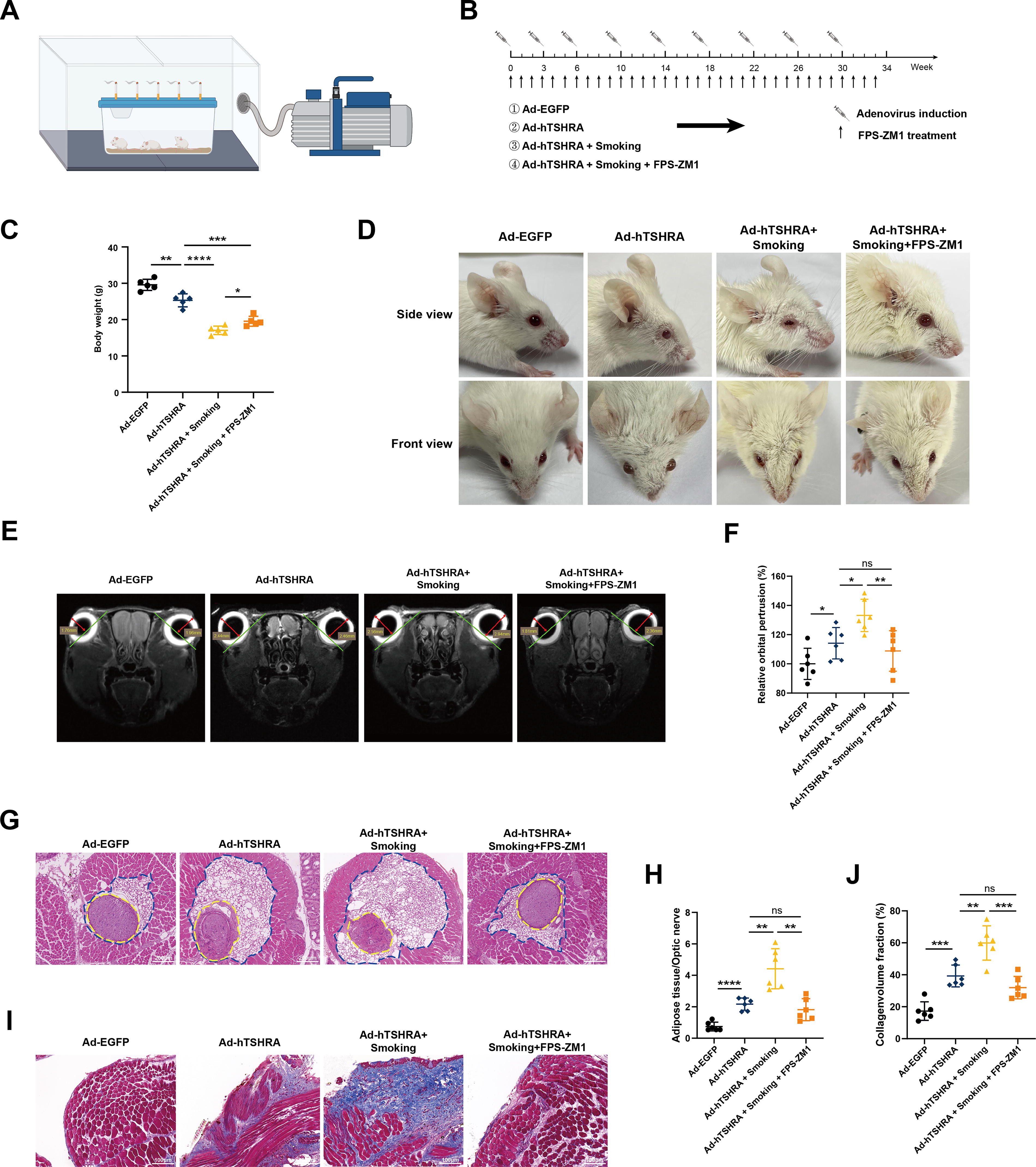
RAGE inhibitor significantly alleviates smoking-induced disease progression in TED mouse model.
Discussion
Cigarette smoking is recognized as an important risk factor for the development of TED, 7,30 with detailed mechanisms of smoking-mediated pathological changes remain undefined. Although smoking is reportedly associated with higher levels of oxidative stress in patients with TED, 12,31 the relationship between the two is unclear. Here, we demonstrated that CSE significantly activated RAGE signaling in TED-derived OFs, leading to increased ROS production. Our study is also the first to reveal that smoking is a driver of orbital inflammation. We confirmed both in vitro and in vivo that smoking resulted in robust inflammatory responses, which correlated with the activation of NF-κB downstream of the RAGE-ROS axis. 22
RAGE-related oxidative stress plays important roles in the fibrogenic processes of several inflammatory diseases, including diabetes and pulmonary fibrosis. 32,33 AGE-RAGE signaling has also been implicated in adipogenic pathophysiology, 34 particularly the enlargement of retro-orbital adipose tissues in patients with TED. 35 Here, we showed that disrupting RAGE function efficiently inhibited fibrosis and adipogenesis aggravated by smoking both in vivo and in vitro. Our work provides further evidence of the link between RAGE and these tissue-remodeling processes in TED, which holds promise as a therapeutic target.
AGE-RAGE signaling has been implicated in the onset of many human inflammatory diseases and, among the diverse ligands that activate RAGE, cigarette smoke is regarded as the most important exogenous source of AGE. 34 RAGE recognizes various ligands produced by cigarette smoke, thereby contributing to smoking-induced pathological injuries such as chronic obstructive pulmonary disease. 36 Furthermore, both orbital tissues and PBMCs from patients with TED and a smoking habit exhibited local and systemic activation of RAGE signaling. These findings highlight the potential of RAGE as a target for the management of smoking-relevant disease progression in TED.
In this study, we applied a prolonged protocol to generate TED model mice and evaluated the impact of smoking on TED pathology. Construction of a platform that mimicked the detrimental impact of daily cigarette smoking in mice allowed us to acquire in vivo data on smoking-induced disease progression and the therapeutic effects of RAGE inhibition. 26 –29 Smoking drastically exacerbated orbital inflammation and pathological changes, and was associated with RAGE signaling activation in TED mice. Through multifaceted analyses, including appearance examination, MRI, and pathology, we confirmed that the RAGE inhibitor significantly attenuated symptoms such as eyelid edema and proptosis. Most previous studies reported the appearance of proptosis as the indicator of mouse TED, 25,37 which may be insufficiently precise. Here, we developed an MRI-based strategy to accurately evaluate the extent of proptosis, quantifying the distances between the corneal apex and inner and outer canthal lines of each eye. This approach supported the therapeutic effect of FPS-ZM1 on proptosis in TED mice. While inhibition of RAGE yielded therapeutic benefits for ocular manifestations, its effects on systemic changes such as thyroid function were comparatively limited. These results further substantiate the notion that RAGE signaling plays a specific role in the pathogenesis of orbital disease.
Our study has several limitations. First, as RAGE is a multiligand receptor, we were not able to determine the specific RAGE ligands in the peripheral blood of smoking patients that cause RAGE activation in the orbit. Second, we only investigated the effects of cigarette smoking on OFs, while it is acknowledged that cigarette exposure may impact multiple cell types. Future studies are warranted to explore changes and mechanisms in other relevant cell populations to gain a more comprehensive understanding of smoking-related pathophysiology. Third, our study primarily focused on changes in TED mice before and after cigarette exposure; we did not include a smoking group of control mice, which may limit our ability to fully evaluate the general systemic effects of smoking. Lastly, due to limitations of biological laboratory facilities, we were unable to conduct specialized chemical or toxicological analyses of CSE. We evaluated the biological effects of cigarette smoke as a whole, rather than dissecting the contribution of individual chemical components. In the future, a more detailed chemical characterization of cigarette smoke constituents will help to better elucidate the underlying pathological mechanisms of smoking.
Conclusions
Our findings demonstrated that cigarette smoking had multifaceted impacts on TED, and RAGE activation played a vital role in the pathophysiology of cigarette smoke-induced disease progression (Fig. 7). Our study also provides a framework for studying the impact of cigarette smoke exposure on human diseases, an emerging public health concern.
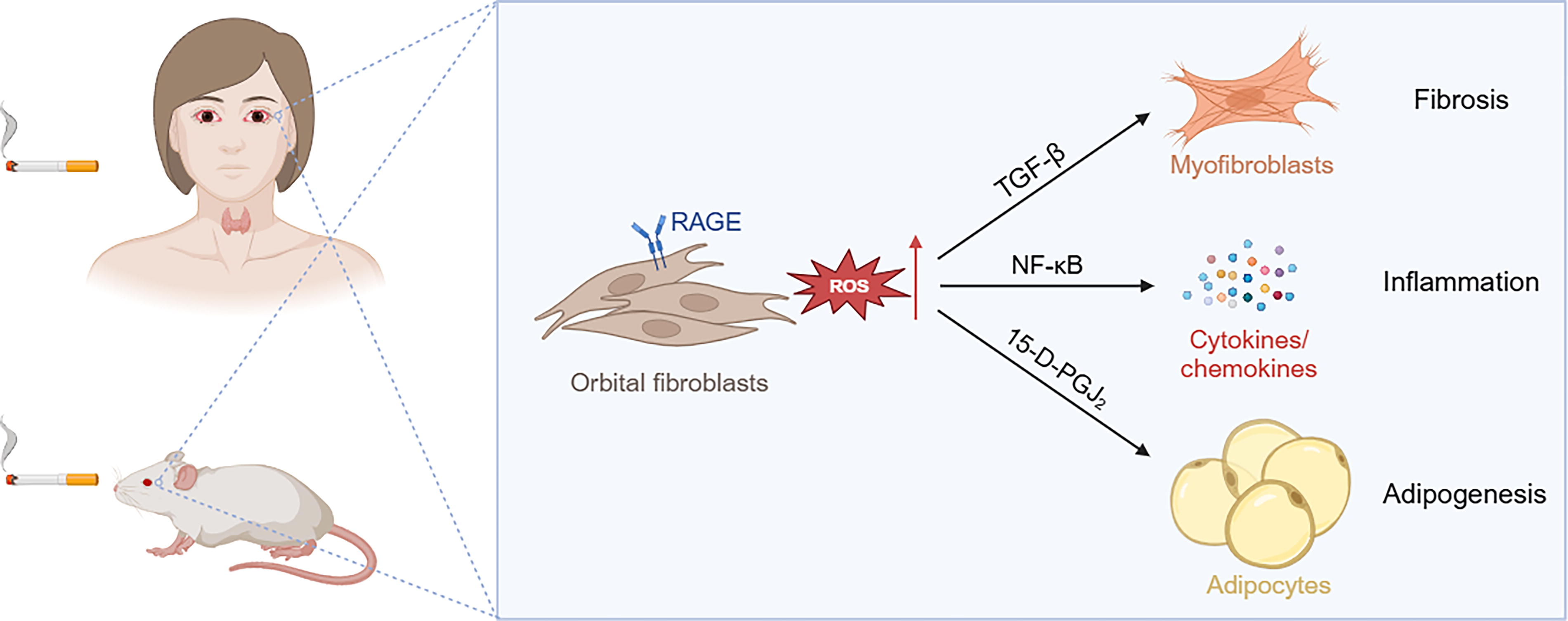
Schematic diagram illustrating the pathological effects of cigarette smoking on thyroid eye disease. This figure was created with BioRender.com.
Footnotes
Authors’ Contributions
J.L.: Conceptualization (supporting), writing—original draft (lead), and formal analysis (lead). T.Z.: Data acquisition (supporting) and writing—original draft (supporting). L.Y.: Writing—original draft (supporting). W.Q.: Software (supporting). L.F.: Methodology (lead). W.Z.: Methodology (supporting). H.Z.: Methodology (supporting). Y.W.: Formal analysis (supporting). B.Y.: Data acquisition (supporting). J.S.: Sample collection (supporting). B.L.: Writing—review and editing (equal). D.L.: Writing—review and editing (equal). Y.L.: Sample collection (lead), histological diagnosis. S.F.: Conceptualization (supporting), writing—review and editing (equal). H.Z.: Conceptualization (lead), writing—review and editing (lead).
Ethics Statement
Orbital tissues were collected from patients with TED who underwent orbital decompression surgery and non-TED healthy controls who underwent blepharoplasty surgery at Shanghai Ninth People’s Hospital. All experiments were conducted in accordance with the ethical guidelines of the Declaration of Helsinki. All participants provided signed informed consent, and the study was approved by the Institutional Ethics Committee, Shanghai Ninth People’s Hospital (SH9H-2021-T371-1). Animal care and use were approved by the Animal Ethics Committee of Shanghai Ninth People’s Hospital (SH9H-2022-A913-1), and complied with institutional and international guidelines.
Author Disclosure Statement
None of the authors has any conflicts of interest to disclose.
Funding Information
This study was supported by the National Natural Science Foundation of China (82071003, 82271122, 82271072, 82241222, 82201166, 82301258, and 82000879), Chang Jiang Scholars Program (T2022065), Shanghai Municipal Commission of Health and Family Planning Project (2022XD006), Shanghai Key Clinical Specialty, Shanghai Eye Disease Research Center (2022ZZ01003), Clinical Acceleration Program of Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine (JYLJ202202), Chenguang Program of Shanghai Education Development Foundation and Shanghai Municipal Education Commission (21CGA18), the Sample Database Project of Shanghai Ninth People’s Hospital (YBKB202211), and the innovative research teams of high-level local universities in Shanghai (SHSMU-ZDCX20210601), Shanghai Municipal Commission of Health and Family Planning (202040117), and projects of the Shanghai Ninth People’s Hospital (JYHX2022001, JYHX2021020).
Supplementary Material
Supplementary Data
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.