
Abstract
Background:
Hantaviral antigens were originally reported more than 20 years ago in tissues of the Eurasian common shrew (Sorex araneus), captured in European and Siberian Russia. The recent discovery of Seewis virus (SWSV) in this soricid species in Switzerland provided an opportunity to investigate its genetic diversity and geographic distribution in Russia.
Methods:
Lung tissues from 45 Eurasian common shrews, 4 Laxmann's shrews (Sorex caecutiens), 3 Siberian large-toothed shrews (Sorex daphaenodon), 9 pygmy shrews (Sorex minutus), 28 tundra shrews (Sorex tundrensis), and 6 Siberian shrews (Crocidura sibirica), captured in 11 localities in Western and Eastern Siberia during June 2007 to September 2008, were analyzed for hantavirus RNA by reverse transcription–polymerase chain reaction.
Results:
Hantavirus L and S segment sequences, detected in 11 S. araneus, 2 S. tundrensis, and 2 S. daphaenodon, were closely related to SWSV, differing from the prototype mp70 strain by 16.3–20.2% at the nucleotide level and 1.4–1.7% at the amino acid level. Alignment and comparison of nucleotide and amino acid sequences showed an intrastrain difference of 0–11.0% and 0% for the L segment and 0.2–8.5% and 0% for the S segment, respectively. Phylogenetic analysis, using neighbor-joining, maximum-likelihood, and Bayesian methods, showed geographic-specific clustering of SWSV strains in Western and Eastern Siberia.
Conclusions:
This is the first definitive report of shrew-borne hantaviruses in Siberia, and demonstrates the impressive distribution of SWSV among phylogenetically related Sorex species. Coevolution and local adaptation of SWSV genetic variants in specific chromosomal races of S. araneus may account for their geographic distribution.
Introduction
Until recently, the single exception to the hantavirus–rodent association was Thottapalayam virus (TPMV), a long-unclassified virus isolated from the Asian house shrew (Suncus murinus), captured in 1964 near Vellore in Tamil Nadu, India (Carey et al. 1971, Zeller et al. 1989). However, whether or not TPMV was naturally harbored by a nonrodent host or represented spillover from a rodent reservoir was a continuing source of debate. Whole-genome sequence analysis of TPMV, showing that it forms a unique phylogenetic clade, has finally put this argument to rest (Song et al. 2007a, Yadav et al. 2007). Also, recent identification of genetically divergent hantaviruses in multiple species of shrews (Order Soricomorpha, Family Soricidae) from widely separated geographic regions provides convincing evidence that soricids serve as reservoir hosts (Arai et al. 2007, 2008a, Klempa et al. 2007, Song et al. 2007b, 2007c, 2009). Moreover, the natural host range has been further expanded with the detection of hantaviruses in moles (family Talpidae), suggesting that ancestral soricomorphs may have served as the early or original mammalian hosts of hantaviruses (Arai et al. 2008b, Kang et al. 2009a, 2009b).
Although hantaviral antigens have been previously reported in the Eurasian common shrew (Sorex araneus), pygmy shrew (Sorex minutus), and Eurasian water shrew (Neomys fodiens), captured in European Russia (Gavrilovskaya et al. 1983, Tkachenko et al. 1983) and Siberia (Miasnikov et al. 1992), definitive molecular evidence of soricid-borne hantaviruses from Russia is lacking. The recent detection of a hantavirus, named Seewis virus (SWSV), in the Eurasian common shrew from Switzerland (Song et al. 2007b) provided an opportunity to investigate the genetic diversity and geographic distribution of SWSV and other hantaviruses harbored by shrews in Western and Eastern Siberia. In this study, we demonstrate the geographic-specific genetic variation of SWSV strains in five administrative regions in Siberia.
Overall, the segregation of SWSV genetic variants according to their geographic locale resembles that of rodent-borne hantaviruses, suggesting long-standing phylogeographic relationships.
Materials and Methods
Trapping and sample collection
Multiple trapping expeditions were conducted in different ecotones, in the seven administrative regions of Western and Eastern Siberia (Altai Republic, Altai, and Krasnoyarsk Krais, and Novosibirsk, Omsk, Kemerovo, and Irkutsk Oblasts) between June 2007 and September 2008 (Table 1 and Fig. 1). Shrews were trapped and tissues were processed according to well-established protocols and safety recommendations (Mills et al. 1995). Sherman traps were set at 10-m intervals and baited with rye bread soaked with sunflower oil. Pitfall traps were spaced at approximately 5-m intervals along 50-m trap lines. All traps were checked twice daily. Species, sex, age class, body mass, and point of capture of each trapped shrew were recorded. Lung tissues were dissected aseptically and stored in liquid nitrogen or RNAlater RNA Stabilization Reagent (Qiagen), for subsequent analysis by reverse transcription–polymerase chain reaction (RT-PCR).
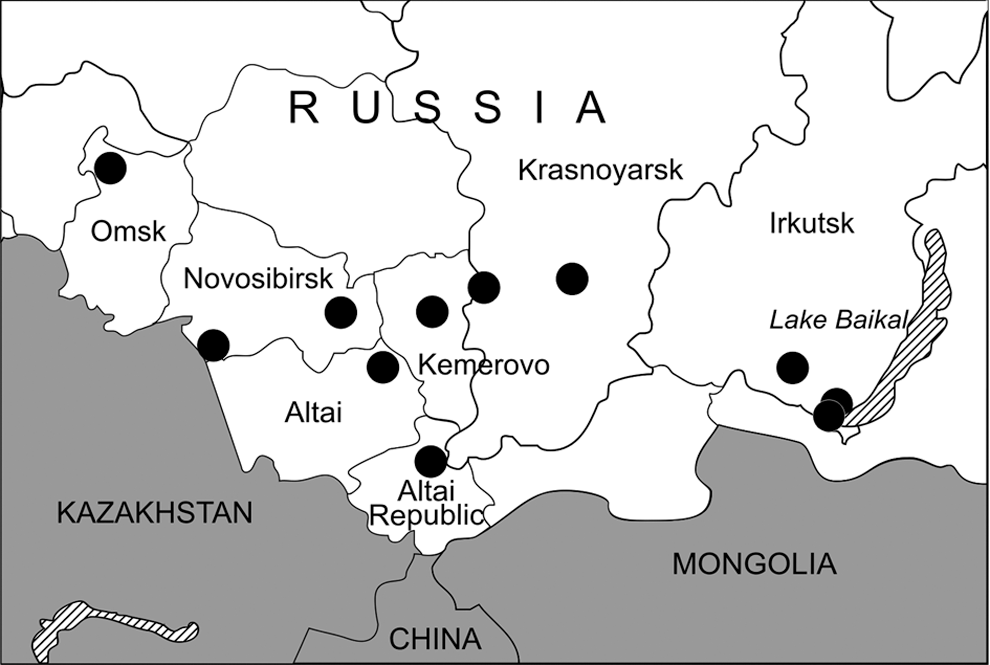
Map of Western and Eastern Siberia showing the administrative regions where shrews were trapped between June 2007 and September 2008.
RT-PCR and DNA sequencing
Total RNA was extracted from tissues using the RNeasy MiniKit (Qiagen), and cDNA was synthesized using Expand reverse transcriptase (Roche) and universal primer 5′-TAGTAGTAGACTCC-3′. All samples were tested by nested RT-PCR (Arai et al. 2008a), using two sets of primers to recover partial large (L) and small (S) segment sequences: L (outer: 5′-ATGTAYGTBAGTGCWGATGC-3′ and 5′-AACCADTCWGTYCCRTCATC-3′; inner: 5′-TGCWGATGCHACIAARTGGTC-3′ and 5′-GCRTCRTCWGARTGRTGDGCA-3′); S (outer: 5′-TAGTAGTAKRCTCCCTAAARAG-3′; inner: 5′-GWGGHCARACWGCAGAYTGG-3′ and 5′-AGCTCAGGATCCATGTCATC-3′).
Sequence and phylogenetic analyses
Hantavirus nucleotide sequences were aligned and compared using ClustalW (Thompson et al. 1994). The distance-based neighbor-joining method supported by MEGA 4.1 (Tamura et al. 2007), as well as maximum-likelihood (ML) and Bayesian methods, implemented in PAUP* (Phylogenetic Analysis Using Parsimony, 4.0b10) (Swofford 2003), RAxML Blackbox web-server (Stamatakis et al. 2008), and MrBayes 3.1 (Ronquist and Huelsenbeck 2003), under the best-fit GTR+I+Γ model of evolution, were used for constructing phylogenetic trees. ML topologies were evaluated by bootstrap analysis of 1000 neighbor-joining iterations in PAUP* or 1000 ML iterations in RAxML. Bayesian analysis consisted of at least 2 million Markov Chain Monte Carlo generations to ensure convergence across two runs of four chains each, with average standard deviations of split frequencies less than 0.01 and effective sample sizes well over 100, resulting in consensus trees supported by posterior-node probabilities (Kang et al. 2009b). GenBank accession numbers for new hantaviral genomic sequences are shown in Table 2.
mtDNA sequencing and host phylogeny
To verify the taxonomic identity of shrews and to ascertain their phylogenetic relationships, genomic DNA was extracted from frozen lung tissue using the QIAamp DNA Mini Kit (Qiagen), and the partial 426-nucleotide region of the cytochrome b gene of mtDNA was amplified by PCR. Amplicons were gel purified with QIAquick Gel Extraction kit (Qiagen) and directly sequenced using ABI Prizm BigDye Terminator kit (PE Applied Biosystem) and an automatic ABI Prism 310 genetic analyzer.
Results and Discussion
Lung tissues from 95 shrews—45 Eurasian common shrews, 4 Laxmann's shrews (Sorex caecutiens), 3 Siberian large-toothed shrews (Sorex daphaenodon), 9 pygmy shrews (S. minutus), 28 tundra shrews (Sorex tundrensis), and 6 Siberian shrews (Crocidura sibirica)—were examined by nested RT-PCR. Hantaviral RNA was detected in 15 shrews captured in 7 of the 11 localities studied: Teletskoye Lake (Altai Republic), Krapivino (Kemerovo Oblast), Karasuk and the suburbs of Novosibirsk City (Novosibirsk Oblast), Parnaya and East Sayan (Krasnoyarsk Krai), and the suburbs of Irkutsk City (Irkutsk Oblast) (Fig. 1 and Table 2). Nucleotide sequence analysis of partial hantavirus L (positions 2968–3313) and S (positions 407–1243) segments from 11 S. araneus, 2 S. tundrensis, and 2 Siberian large-toothed shrew (Table 2) showed close similarity to SWSV, previously reported from S. araneus captured in Switzerland (Song et al. 2007b). The sequences appeared to be genetic variants of SWSV, differing from the prototype mp70 strain by 16.3–20.2% at the nucleotide level and 1.7% at the amino acid level for the L segment, and by 17.4–19.1% and 1.4% for the S segment, respectively. Pair-wise alignment and comparison of nucleotide and amino acid sequences showed an intrastrain difference of 0–11.0% and 0% for the L segment and 0.2–8.5% and 0% for the S segment, respectively. Nucleotide divergence from other previously known shrew- and rodent-borne hantavirus strains ranged from 24.5% to 35.6% and from 31.9% to 48.4%, respectively.
The new sequences showed geographic-specific clustering. Nucleotide sequences in the same locality demonstrated minimal differences even if they were recovered from different soricid hosts. For example, hantavirus sequences recovered from S. araneus and S. tundrensis captured near Karasuk village in Novosibirsk Oblast were identical for the L segment and showed 0.2% difference for the S segment. Similarly, S and L segment nucleotide sequences from S. daphaenodon and S. tundrensis captured in Irkutsk Oblast differed by 0.3–0.6% and 0.9–1.2%, respectively. By contrast, SWSV sequences from five S. araneus captured near Teletskoye Lake in Altai Republic formed two separate groups and showed high intergroup L segment nucleotide sequence divergence (6.4–7.7%). A lower divergence was demonstrated for S segment sequences (0.9–2.4%).
Phylogenetic analysis, based on 346 and 837 nucleotides of the L and S segments, respectively, using the ML method, implemented in RAxML Blackbox web-server showed geographic-specific clustering of SWSV strains (Fig. 2), not unlike that reported previously for rodent-borne hantaviruses, including Hantaan virus in the striped field mouse (Song et al. 2000), Puumala virus in the bank vole (Plyusnin et al. 1994b, 1995), Muju virus in the royal vole (Song et al. 2007), and Tula virus in the European common vole (Song et al. 2004). Similar topologies were evident using neighbor-joining and Bayesian methods. The correlation between hantavirus genetic variation and geographic origin of the reservoir species provided support for local host-specific adaptation through deep evolutionary time.

Phylogenetic trees, based on 837- and 346-nucleotide regions of the S and L segments, respectively, generated using the maximum-likelihood method, implemented in RAxML Blackbox web-server, under the best-fit GTR+I+Γ model of evolution, showing geographic-specific clustering of Seewis (SWS) virus strains. Bootstrap confidence limits were based on 1000 replicates. Phylogenetic positions of SWS virus strains from Siberia are shown in relationship to previously reported rodent- and soricid-hantaviruses, including SWS virus (SWS mp70, EF636024, EF636026), Ash River virus (ARR MSB73418, EF650086, EF619961), Jemez Springs virus (JMS MSB89332, FJ593499, FJ593501), Cao Bang virus (CBN CBN-3, EF543524, EF543525), Thottapalayam virus (TPM VRC66412, AY526097, EU001330), Hantaan virus (HTN 76-118, X55901, M14626), Dobrava virus (DOB Greece, AJ410619, AJ410617), Seoul virus (SEO 80-39, AY273791, X56492), Tula virus (TUL 5302v, NC_005227, AJ005637), Puumala virus (PUU Sotkamo, X61035, Z66548), and Sin Nombre virus (SN NMH10, L25784, L37902).
As verified by phylogenetic analysis of the partial 426-nucleotide region of the cytochrome b gene, SWSV-positive shrews were identified as S. araneus, S. tundrensis, and S. daphaenodon (GenBank accession numbers GQ355606–GQ355620). Unlike the hantavirus sequences, the host mtDNA sequences did not segregate into distinct lineages, according to the geographic locales or capture sites of the shrews.
Previous studies of shrew communities, conducted over multiple years in four of the five administrative regions, where hantavirus-positive shrews were detected, showed that S. araneus was the predominant species, comprising more then 30% of the shrew population: 49% in Teletskoye Lake, 32% in Karasuk, 71% in Novosibirsk, and 47% in Kemerovo. As determined by RT-PCR, the prevalence of hantavirus infection among Eurasian common shrews in each locality varied considerably, ranging from 2 of 2 (100%) in East Sayan, 5 of 9 (55.5%) in Teletskoye Lake, 1 of 3 (33.3%) in Parnaya, 1 of 6 (16.7%) in suburbs of Novosibirsk City and Krapivino, and 1 of 13 (7.7%) in Karasuk. We were unable to find a significant correlation between the infection rate and population density or male/female ratio in these sites. In our study, high S. araneus density was registered near Teletskoye Lake (13.6 individuals per 100 trap-days; male/female = 3/6), in Krapivino (10.7 individuals/100 trap-days; male/female = 2/4), and in Novosibirsk City (16.9 individuals/100 trap-days; male/female = 4/2), compared to low S. araneus density in Karasuk (3.1 individuals/100 trap-days; male/female = 5/2).
The Eurasian common shrew is among the most widely dispersed small mammal species in Eurasia. S. araneus has one of the most variable karyotypes among small mammals, displaying 70 chromosome races throughout its distribution in Europe and Siberia. The seven chromosome races in Siberia sequentially replaced each other in the latitudinal direction (Polyakov et al. 2001). Based on previous studies we suggest that at least four chromosomal races of the Eurasian common shrew are present in areas covered by our research: Novosibirsk race (Novosib-Sa374), Tomsk race (Kemerovo-Sa65 and Parnaya-Sa1220), Strelka race (Krasn-Sa5 and Krasn-Sa6), and Altai race (Telet-Sa300, Telet-Sa301, Telet-Sa313, Telet-Sa321, and Telet-Sa500). Our data demonstrated high nucleotide L-sequence divergence (up to 7.7%) among sequences from Teletskoye Lake. Based on the hypothesis of coevolution of hantaviruses and their hosts, we suggest that two separate SWSV sublineages had different evolution histories and, most probably, circulated not in a single Altai race, but rather in two different races. A second race may be a new race or the Seminsk race that was described previously in the Altai Republic in a site located 100 km away from the capture site near Teletskoye Lake (Polyakov et al. 2001).
This report represents the first molecular evidence of shrew-borne hantaviruses in Siberia. Our data corroborate earlier reports of hantaviral antigens in S. araneus captured in Russia (Gavrilovskaya et al. 1983, Tkachenko et al. 1983, Miasnikov et al. 1992). Also, together with recent findings of the geographic-specific genetic variation of SWSV in S. araneus captured in Finland and Hungary (H.J. Kang and R. Yanagihara, unpublished observations), as well as in Germany and Austria (R. Dürrwald and N. Nowotny, pers. comm.), our data establish the widespread circulation of SWSV throughout the geographic range of S. araneus, spanning across Northern Europe to Eastern Siberia. Moreover, detection of SWSV in S. araneus, S. tundrensis, and S. daphaenodon in Siberia is consistent with the shared ancestry of these closely related Sorex species within the S. araneus group (Searle and Wójcik 1998). The vast distribution of SWSV, therefore, may be a consequence of a primordial hantavirus infecting ancestral Sorex shrews, with subsequent host switching and local host adaptation through deep evolutionary time. Intensified studies, now underway, in search of SWSV and other genetically distinct hantaviruses in sympatric and syntopic shrew species across Siberia will provide additional important insights into the complex phylogeographic history of hantaviruses.
A pressing issue is whether nonrodent-borne hantaviruses cause infection or disease in humans. Although suggestive evidence of TPMV infection in humans has been reported (Okumura et al. 2007), neither TPMV nor any of the newly identified soricid- and talpid-borne hantaviruses has been etiologically linked to specific diseases or syndromes in humans. Nevertheless, it is still far too premature to label these viruses as being nonpathogenic. Instead, the realization that rodent-borne hantaviruses are capable of causing diseases as clinically disparate as HFRS and HCPS raises the possibility that still-orphan hantaviruses harbored by shrews and moles might similarly cause diseases that do not clinically resemble either HFRS or HCPS. Efforts are now underway to develop recombinant nucleocapsid proteins and other diagnostic reagents to test sera from human patients with various diseases for antibodies against these newfound hantaviruses. In addition, sera from mammalogists and field biologists having exposure to soricomorphs are being analyzed.
Footnotes
Acknowledgments
We thank our colleagues, Dr. Alexander Timoshenko, Dr. Dmitry Petrovski, and Dr. Alexander Pozdnyakov, for providing additional shrew tissues for testing. This work was supported in part by grants from the Biotechnology Engagement Program, through the International Science and Technology Center (#0805.2), and from the National Institute of Allergy and Infectious Diseases, National Institutes of Health (R01AI075057).
Disclosure Statement
All authors have no competing financial interests. The funding agency had no role in study design, data collection and analysis, decision to publish, or preparation of the article.