
Abstract
Genetically distinct hantaviruses have been identified recently in multiple species of shrews (Order Soricomorpha, Family Soricidae) in Eurasia and North America. To corroborate decades-old reports of hantaviral antigens in shrews from Russia, archival liver and lung tissues from 4 Siberian large-toothed shrews (Sorex daphaenodon), 5 Eurasian least shrews (Sorex minutissimus), 12 flat-skulled shrews (Sorex roboratus), and 18 tundra shrews (Sorex tundrensis), captured in the Sakha Republic in northeastern Siberia during July and August 2006, were analyzed for hantavirus RNA by reverse transcription–polymerase chain reaction. A novel hantavirus, named Kenkeme virus, was detected in a flat-skulled shrew. Sequence analysis of the full-length S and partial M and L segments indicated that Kenkeme virus was genetically and phylogenetically distinct from Seewis virus harbored by the Eurasian common shrew (Sorex araneus), as well as all other rodent-, soricid-, and talpid-borne hantaviruses.
Newly identified shrew-borne hantaviruses include Tanganya virus in the Therese's shrew (Crocidura theresae) (Klempa et al. 2007), Camp Ripley virus (RPLV) in the northern short-tailed shrew (Blarina brevicauda) (Arai et al. 2007), Cao Bang virus (CBNV) in the Chinese mole shrew (Anourosorex squamipes) (Song et al. 2007b), Seewis virus (SWSV) in the Eurasian common shrew (Sorex araneus) (Song et al. 2007a), Ash River virus (ARRV) in the masked shrew (Sorex cinereus) (Arai et al. 2008a), Jemez Springs virus (JMSV) in the dusky shrew (Sorex monticolus) (Arai et al. 2008a), and Imjin virus (MJNV) in the Ussuri white-toothed shrew (Crocidura lasiura) (Song et al. 2009). In addition, phylogenetically distinct hantaviruses have been identified in moles (Family Talpidae), including Asama virus in the Japanese shrew mole (Urotrichus talpoides) (Arai et al. 2008b), Oxbow virus in the American shrew mole (Neurotrichus gibbsii) (Kang et al. 2009b), and Nova virus in the European common mole (Talpa europaea) (Kang et al. 2009c).
In an attempt to corroborate decades-old reports of hantaviral antigens in shrews from Russia (Gavrilovskaya et al. 1983, Tkachenko et al. 1983, Miasnikov et al. 1992), archival liver or lung tissues from 4 Siberian large-toothed shrews (Sorex daphaenodon), 5 Eurasian least shrews (Sorex minutissimus), 12 flat-skulled shrews (Sorex roboratus), and 18 tundra shrews (Sorex tundrensis), collected as part of the Beringian Coevolution Project in the Sakha Republic in northeastern Siberia (Fig. 1) during July and August 2006, were analyzed for hantavirus RNA by reverse transcription–polymerase chain reaction (PCR), using primers designed from highly conserved regions of hantavirus genomes (Arai et al. 2008a, Kang et al. 2009b, 2009c). cDNA was synthesized using the SuperScript III First-Strand Synthesis System (Invitrogen, San Diego, CA) and an oligonucleotide primer (OSM55, 5′-TAGTAGTAGACTCC-3′) designed from the conserved 3′-end of the S, M, and L segments of hantaviruses. First- and second-round PCRs were performed in 20 μL reaction mixtures containing 250 μM dNTP, 2 mM MgCl2, 1 U AmpliTaq polymerase (Roche, Basel, Switzerland), and 0.25 μM of each primer. Initial denaturation at 94°C for 5 min was followed by two cycles each of denaturation at 94°C for 40 s, two-degree step-down annealing from 48°C to 38°C for 40 s, and elongation at 72°C for 1 min, then 32 cycles of denaturation at 94°C for 40 s, annealing at 42°C for 40 s, and elongation at 72°C for 1 min, in a GeneAmp PCR 9700 thermal cycler (Perkin-Elmer, Waltham, MA). Amplicons were separated by electrophoresis on 1.5% agarose gels and purified using the QIAQuick Gel Extraction Kit (Qiagen, Hilden, Germany). DNA was sequenced directly using an ABI Prism 377XL Genetic Analyzer (Applied Biosystems, Foster City, CA). Several amplicons from the same region were sequenced and sequences were cleaned using the Lasergene program version 5 (DNASTAR, Madison, WI).
Map of Russia showing the Sakha Republic in Eastern Siberia, where trapping of shrews was conducted as part of the Beringian Coevolution Project during July and August 2006.
All but one sample was negative for hantavirus RNA. The single exception, designated Kenkeme virus (KKMV) strain MSB148794, was a hantavirus detected in a flat-skulled shrew, captured near the Kenkeme River, 40 km west of Yakutsk (62.07003°, 128.93831°). The taxonomic identity of this shrew was confirmed by analysis of the entire 1140-bp cytochrome b mtDNA sequence.
The full-length 1640-nucleotide S-genomic segment of KKMV MSB148794 contained a single open reading frame, encoding a predicted nucleocapsid (N) protein of 429 amino acids (nucleotide positions 48 to 1337), and 47- and 303-nucleotide 3′- and 5′-noncoding regions, respectively. The predicted secondary structure of the KKMV N protein, as determined by various software available in the NPS@ structure server (
Pairwise alignment and comparison of the full-length KKMV S segment, using ClustalW, exhibited uniformly low nucleotide sequence similarity to representative rodent-associated hantaviruses, ranging from 61.6% for Hantaan virus strain 76–118 and 60.2% for Dobrava virus strain Greece, and 59.3–60.9% for hantaviruses harbored by arvicolid and neotomine/sigmodontine rodents (Table 1). At the amino acid level, the KKMV N protein sequence was most similar to but distinct from that of SWSV, differing by 16.3%. Otherwise, the KKMV N protein differed by 38.1–41.9% and 30.5–55.1%, respectively, from representative rodent- and soricomorph-associated hantaviruses, supporting the notion that it constitutes a distinct hantavirus species, based on criteria set forth by the International Committee for Taxonomy of Viruses (Fauquet et al. 2005) and a more recent exhaustive analysis of hantavirus genomes (Maes et al. 2009).
ANDV, Andes virus; ARRV, Ash River virus; ASAV, Asama virus; CBNV, Cao Bang virus; DOBV, Dobrava virus; HTNV, Hantaan virus; JMSV, Jemez Spring virus; KKMV, Kenkeme virus; MJNV, Imjin virus; NVAV, Nova virus; OXBV, Oxbow virus; PHV, Prospect Hill virus; PUUV, Puumala virus; RPLV, Camp Ripley virus; SEOV, Seoul virus; SNV, Sin Nombre virus; SOOV, Soochong virus; SWSV, Seewis virus; TPMV, Thottapalayam virus; TULV, Tula virus; nt, nucleotides; aa, amino acids.
Similarly, low but wider degrees of sequence homology, ranging from 54.9% to 74.4% and 61.4% to 77.3%, respectively, were found in comparing the partial 1002-nucleotide M and 4295-nucleotide L segments of KKMV with other shrew-borne hantaviruses, including ARRV, CBNV, JMSV, SWSV, RPLV, MJNV, and TPMV (Table 1). Differences in the deduced amino acid sequences of the envelope glycoprotein and viral RNA-dependent RNA polymerase also supported KKMV MSB148794 as being a genetically distinct hantavirus.
Phylogenetic trees, based on the S-, M-, and L-segment sequences, generated by maximum likelihood and Bayesian methods, implemented in PAUP* (Phylogenetic Analysis Using Parsimony, 4.0b10) (Swofford 2003), RAxML Blackbox webserver (Stamatakis et al. 2008), and MrBayes 3.1 (Ronquist et al. 2003), under the best-fit GTR+I+Γ model of evolution established using jModeltest 0.1.1 (Posada 2008), revealed similar topologies, which were well supported by bootstrap analysis (Fig. 2). KKMV and SWSV occupied separate lineages, in keeping with the phylogenetic relationship between their respective soricid reservoir hosts, S. roboratus and S. araneus. That is, S. roboratus, an oligochaete-eating soricine shrew usually residing in flood-plain habitats, belongs to the Sorex caecutiens group, an evolutionary clade of Palearctic shrews, which is distinct from the S. araneus group.
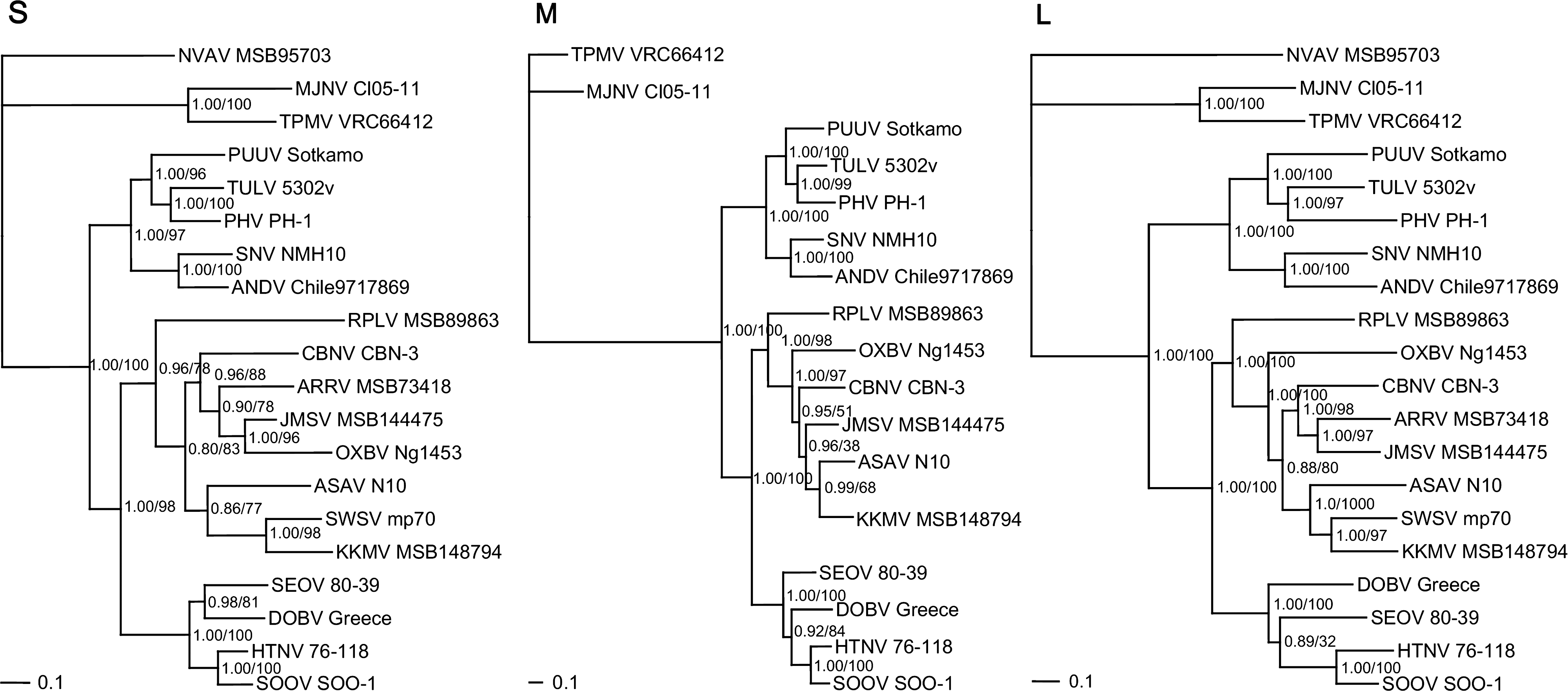
Phylogenetic trees generated by the Bayesian method, under the best-fit GTR+I+Γ model of evolution, based on the full-length S and partial M and L genomic segments of Kenkeme virus (KKMV MSB148794) and other selected hantaviruses. The phylogenetic positions of KKMV are shown in relationship to Cao Bang virus (CBNV CBN-3; EF543524, EF543526, EF543525) from the Chinese mole shrew (Anourosorex squamipes), Camp Ripley virus (RPLV MSB89863; FJ790772, EF540774, EF540771) from the northern short-tailed shrew (Blarina brevicauda), Seewis virus (SWSV mp70; EF636024, EF636026) from the Eurasian common shrew (Sorex araneus), Ash River virus (ARRV MSB73418; EF650086, EF619961) from the masked shrew (Sorex cinereus), Jemez Spring virus (JMSV MSB144475; FJ593499, FJ593500, FJ593501) from the dusky shrew (Sorex monticolus), Imjin virus (MJNV Cl05-11; EF641804, EF641798, EF641806) from the Ussuri white-toothed shrew (Crocidura lasiura), Thottapalayam virus (TPMV VRC66412; AY526097, EU001329, EU001330) from the Asian house shrew (Suncus murinus), Asama virus (ASAV N10; EU929072, EU929075, EU929078) from the Japanese shrew mole (Urotrichus talpoides), Oxbow virus (OXBV Ng1453; FJ539166, FJ539167, FJ593497) from the American shrew mole (Neurotrichus gibbsii), and Nova virus (NVAV MSB95703; FJ539168, FJ593498) from the European common mole (Talpa europaea). Also shown are representative rodent-borne hantaviruses, including Hantaan virus (HTNV 76–118; NC_005218, Y00386, NC_005222), Soochong virus (SOOV SOO-1; AY675349, AY675353, DQ056292), Dobrava virus (DOBV Greece; NC_005233, NC_005234, NC_005235), Seoul virus (SEOV 80-39; NC_005236, NC_005237, NC_005238), Tula virus (TULV 5302v; NC_005227, NC_005228, NC_005226), Puumala virus (PUUV Sotkamo; NC_005224, NC_005223, NC_005225), Prospect Hill virus (PHV PH-1; Z49098, X55129, EF646763), Sin Nombre virus (SNV NMH10; NC_005216, NC_005215, NC_005217), and Andes virus (ANDV Chile9717869; NC_003466, NC_003467, NC_003468). GenBank accession numbers are GQ306148, GQ306149, and GQ306150 for the S, M, and L segments of KKMV MSB148794, respectively. SWSV, ARRV, and NVAV are not shown in the M-segment tree because sequences were unavailable for analysis. The numbers at each node are posterior node probabilities (left of slash) based on 30,000 trees, and maximum-likelihood (ML) bootstrap values (right of slash) based on 1000 bootstrap replicates. Bayesian analysis consisted of two replicate Markov Chain Monte Carlo runs of four chains of 2 million generations, each sampled every 100 generations with a burn-in of 5000 (25%). The scale bar indicates nucleotide substitutions per site.
SWSV, originally identified in the Eurasian common shrew from Switzerland (Song et al. 2007a), has recently been detected in this shrew species in Finland and Hungary (Kang et al. 2009a) and in Germany and Austria (R. Dürrwald and N. Nowotny, personal communication), as well as in Western and Eastern Siberia (Yashina et al. 2009). In addition, SWSV genetic variants have been found in the tundra shrew and Siberian large-toothed shrew in Siberia (Yashina et al. 2009). To our knowledge, however, KKMV represents the first evidence of a genetically distinct hantavirus in a previously unrecognized soricid species from Russia. Nevertheless, in-depth studies are warranted to establish if the flat-skulled shrew is truly the principal reservoir host of KKMV, because S. roboratus is sympatric or parapatric with 12 other Sorex species throughout its geographic range in western and central Russia and Mongolia (Sheftel 2005) and so opportunities exist for cross-species transmission. Investigations to ascertain if genetic variants of KKMV circulate in such sympatric shrew species are now underway.
The discovery of KKMV adds to the rich diversity of recently identified hantaviruses in shrews and lends support to the emerging paradigm-shifting concept that ancestral soricomorphs, rather than rodents, may have served as the early mammalian hosts of primordial hantaviruses. That is, in keeping with all other present-day members of the Bunyaviridae, insects or arthropods may have originally served as vectors of ancient hantaviruses, which became established in soricomorphs. When viewed within this hypothetical context, the evolutionary history of hantaviruses is far more ancient and complex than previously imagined. Although host switching with subsequent host-specific adaptations has occurred during hantavirus evolution (Vapalahti et al. 1999, Nemirov et al. 2002), these events alone do not satisfactorily explain the coexistence and distribution of genetically distinct hantaviruses among species in two taxonomic orders of small mammals spanning four continents (Arai et al. 2008b, Kang et al. 2009b, 2009c).
Many rodent-borne hantaviruses in Eurasia and the Americas appear to be nonpathogenic. Similarly, none of the recently identified shrew-borne hantaviruses has hitherto been shown to cause diseases in humans. However, serological evidence of TPMV infection has recently been reported in a febrile patient (Okumura et al. 2007), suggesting that human infection with other soricid-associated hantaviruses may be possible. Intensive efforts are currently underway in search of shrew-borne hantavirus infection in patients with febrile illnesses and other ill-defined syndromes. Also, as flat-skulled shrews inhabit taiga and meadows in tundra habitats, individuals, particularly mammalogists, forestry workers, and hunters, who venture into such ecotopes, may be at increased risk of exposure to KKMV.
Footnotes
Acknowledgments
This work was supported in part by the U.S. Public Service grants R01AI075057 from the National Institute of Allergy and Infectious Diseases, and P20RR018727 (Centers of Biomedical Research Excellence) and G12RR003061 (Research Centers in Minority Institutions) from the National Center for Research Resources, National Institutes of Health. Shrew samples were collected through grants DEB0196095 and DEB9981915 from the National Science Foundation.
Disclosure Statement
All authors have no competing financial interests. The funding agency had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.