
Abstract
Tick-borne relapsing fever (TBRF) is endemic in Africa and Eurasia and attributed to different Borrelia species. In the Central Asia and Middle Eastern countries, TBRF is caused mainly by Borrelia persica; however, other Borrelia species such B. microtti, B. latyschewii, B. baltazardi, and B. caucasica have also been described. The classic taxonomy of Borrelia spp. is based on the cospeciation concept that is very complex and rather confusing. In this study, we report two DNA-based methods to discriminate B. persica and B. microtti, the two main prevalent species in the region. Molecular typing of the species was performed using (i) restriction fragment length polymorphism analysis of polymerase chain reaction (PCR)-amplified fragments of either 16S-rRNA or glpQ genes, and (ii) species-specific PCR of glpQ gene. Sequence analyses of the data obtained in this study indicate that the glpQ gene is more variable than 16S-rRNA (6.9% vs. 1.2%); thus glpQ is a more useful marker for discrimination of B. persica from B. microtti. The 16S-rRNA fragment comprises only one useful species-specific restriction site (TaqI), whereas the glpQ fragment includes several species-specific restriction sites and its digestion by DraI, TaqI, EcoRV, HinfI, or SspI results in distinctively different PCR–restriction fragment length polymorphism patterns for the two species. Further, the species-specific primers amplified a 253-bp fragment for B. persica and a 451-bp one for B. microtti. Phylogenetic analysis of the data revealed that B. microtti and B. persica are associated to the African and new world RF agents, respectively. This study demonstrates that both typing methods are simple, sensitive, and fast, and that they allow one to differentiate between B. persica and B. microtti. This could prove that both methods are important and useful in monitoring of TBRF disease in endemic areas.
Introduction
In Asia, TBRF is the most common tick-borne human disease and results mainly from an infection with the spirochete Borrelia persica. The disease was first described by Dschunkowsky (1913) in the Ardabil region of Iran, and the agent responsible for it was called B. persica (Euzeby 1997). Since then, several cases of RF have been reported all over the region, attributed mainly to B. persica. Other bacteria such as Borrelia microtii/microtti, Borrelia latyschewii/latshywii, Borrelia baltazardi/bartarzardi, and Borrelia caucasica have also been described (Karimi et al. 1979, Barbour and Hayes 1986, Euzeby 1997, Assous and Wilamowski 2009). However, B. persica is the most important and prevalent agent of TBRF in humans (Assous and Wilamowski 2009). The tick Ornithodoros tholozani is the main vector of B. persica and its distribution overlaps the distribution area of clinical RF cases caused by B. persica. O. tholozani is widely distributed throughout Indian subcontinent, the southern countries of the former USSR (Kazakhstan, Kirgizia, Tajikistan, Turkmenistan, and Uzbekistan), and the Middle East (Goubau 1984, Parola and Raoult 2001). Reservoir hosts are usually wild rodents (Rebaudet and Parola 2006). Following B. persica, the next prevalent species in the region, particularly in Iran, is B. microtti (Bahrmand et al. 1996, Assmar et al. 2002, Masoumi Asl et al. 2009). This species was discovered by the Iranian scientist Rafiee in 1946 (Assmar et al. 2002). Its vector is the tick Ornithodoros erraticus (small species), which is responsible for its spread from Afghanistan to East Africa (Karimi 1980).
Different Borrelia species cause various clinical features from no symptom to very severe; however, severity also depends on inoculum density and underlying medical conditions. Children and women appear to go through a more intense course of the disease (Barbour 1999). Conventional taxonomy of the Borrelia species has been based on the cospeciation of each species of tick vector and its associated RF-causing Borrelia. In this approach, the only characters that can be used in taxonomy were (i) the vector species, (ii) the geographical location, (iii) the spectrum of laboratory animal pathogenicity, and (iv) the experimental transmission of Borrelia by different arthropods (Assous and Wilamowski 2009). This approach contributes to creating high-level complexity and confusion (Parola and Raoult 2001), and these characters do not allow differentiation between two Borrelia species with sufficient precision.
In Iran, four species B. persica, B. microtti, B. latyschewii, and B. baltazardi have been reported, and each one of these species is responsible for the spread of the disease in certain areas of the country (Karimi 1980, Masoumi Asl et al. 2009). However, there is no recent report on presence of B. latyschewii and B. baltazardi for several years in the country. In Iran, practical confirmation of spirochete infection in humans has relied mainly on microscopic identification of the bacteria in blood smears of febrile patients. Alternatively, laboratory animal inoculation has been used for isolation and diagnosis of Borrelia species. Testing blood smears is limited to just a few days after inoculation of susceptible laboratory animal (adult guinea pig, newborn rabbit, new born rat, and adult white mice). It also requires a highly experienced observer to identify the spirochetes and may lead to a tedious and often unsuccessful microscopic search (Geigy 1968, Karimi et al. 1979).
The development of the polymerase chain reaction (PCR) has offered a new dimension in the diagnosis of infectious diseases. This molecular method, which is used with increasing frequency, offers the possibility of reliable identification of Borrelia species (Fukunaga et al. 1996, Schmidt 1997, Brahim et al. 2005, Halperin et al. 2006, Cerar et al. 2008, Horka et al. 2008). Most of the researches reported so far have been focused on the Borrelia species that cause Lyme disease. However, there is no molecular data available for B. microtti and the utility of PCR for discrimination of B. persica and B. microtti. In this study, partial fragments of glycerophosphodiester phosphodiesterase (glpQ) and 16S ribosomal RNA (16S-rRNA) genes from B. persica and B. microtti were amplified and sequenced, and based on the sequence variation, species-specific PCR and PCR–restriction fragment length polymorphism (RFLP) assays were developed to discriminate between them.
Materials and Methods
Bacteria
B. persica and B. microtti samples were prepared from the Pasteur Institute of Iran. These species were originally isolated from O. tholozani and O. erraticus, respectively, in northwestern Iran. Stocks of these bacteria are usually kept in the laboratory by culturing them in guinea pigs and newborn white mice, respectively. On days 5–10 following inoculation of the bacteria into the animals, blood samples were prepared from the animals and examined by dark-field microscopy for growth of spirochetes. The infected blood samples were then subjected to DNA extraction.
DNA extraction and PCR amplification of 16S-rRNA and glpQ
Genomic DNA from 100 μL of the Borrelia-infected blood samples were extracted with Bioneer Tissue DNA Spin Kit (Bioneer), according to the manufacturer's instructions.
The 16S-rRNA gene was amplified by PCR from the blood of the animals using the primers of Rec4 (5′-ATGCTAGAAACTGCATGA-3′) and Rec9 (5′-TCGTCTGAGTCCCCATCT-3′), as previously described by Ras et al. (1996). These primers are able to amplify a 523-bp PCR product for Borrelia spp. Briefly, 5 μL (5–10 ng) of isolated DNA was used as template for amplification with the primers using initial denaturation of 3 min at 94°C, followed by 35 cycles of 1 min at 93°C, 1 min at 45°C, and 1 min at 72°C, and finally, a 7-min hold at 72°C. PCR was performed in 30 μL reaction volume containing 15 μL of ready-to-use reaction mixture (Bioneer), 10 pmol of each primer, and 5 μL of template DNA extracted from samples.
For glpQ gene PCR, a 668-bp fragment of the gene was successfully amplified for both species B. persica and B. microtti using the primers 128f (5′-CAG AAC ATA CCT TAG AAG CTC AAG C-3′) and 795r (5′-GGG TAT CCA AGG TCC AAT-3′), as previously described by Halperin et al. (2006). PCR was performed in 30 μL reaction volume containing 15 μL of ready-to-use reaction mixture (Bioneer), 1 pmol of each primer, and 3 μL of the template DNA. The PCR protocol included 5 initiation cycles (40 s at 95°C, 50 s at 45°C, and 1 min at 72°C), followed by 25 primary cycles (40 s at 95°C, 50 s at 51°C, and 1 min at 72°C), and 5 min final extension at 72°C.
Amplicons of both PCR sets were visualized on agarose (1%, w/v) gels stained with ethidium bromide. To confirm the identity of the Borrelia species used in this study, a few PCR products of glpQ and 16S-rRNA from both species were picked at random and sequenced (Seqlab). Obtained sequences were deposited in GenBank under accessions nos. EU914141 and EU914143 for B. persica and EU914142 and EU914144 for B. microtti. The sequences were identified at the species level by sequence comparison with the GenBank entries using BLAST and the software for phylogenetic analysis online embedded in Pubmed (
Polymerase chain reaction–restriction fragment length polymorphism
To select appropriate restriction enzymes for discrimination of B. persica from B. microtti, the sequences of 16S-rRNA and glpQ of both species were screened by the Nubcutter (
Primer design and species-specific PCR
Sequences of the glpQ gene of B. persica and B. microtti were aligned by using the Clustal X program (Thompson et al. 1997). This allowed us to identify specific sequence elements for the design of species-specific diagnostic primers. Specific sequences were found on the variable sites of the GlpQ gene and these showed nucleotide identity either with B. persica or B. microtti species where each specific primer had a sequence discrepancy with the other species. The reverse primer BPGLPQR was designed and used with the 128f forward primer to amplify a 252-bp-specific band for B. persica. The specificity of the primer was tested against the strain of B. microtti. Also a forward primer named BMGLPGF was designed and used with the 795r reverse primer to amplify a 457-bp-specific band for B. microtti. The specificity of the primer was tested against the strain of B. persica. Amplification reactions were performed as described earlier for glpQ PCR amplification. The products were analyzed by electrophoresis in 2% (w/v) agarose gels, and product size was estimated with comparison to a DNA ladder (100-bp DNA ladder; Fermentas). Because of lack of proper specific sequences in 16S-rRNA, we were not able to design proper species-specific primers for the species.
Results
PCR analysis of 16S-rDNA and glpQ
The universal Rec4/Rec9 of 16S-rDNA and 128f/795r of glpQ gene primer sets worked successfully for both species. The sizes of PCR products were identical for both species: 523 and 668 bp for the 16S-rDNA and glpQ fragments, respectively. Further, no size variation within the 16S-rDNA or glpQ regions was observed amongst independent isolates from each species.
DNA sequence comparison of 668 bp of glpQ from B. persica and B. microtti showed that there were 44 (6.6%) substitutions between the species. This rate was much lower in 16S-rRNA, where only six (1.2%) substitutions were found within the 520-bp fragment between the two species.
The 16S-rRNA sequence of B. persica obtained in this study was found to be identical to that of the B. persica isolates from Iran (GenBank accession no. BPU42297). However, the glpQ sequence of B. persica obtained in this study differed 3.7% from a haplotype of B. persica from Iran (GenBank accession no. AV530742).
The glpQ sequence of B. microtti obtained in this study was found to be unique and different to that of other Borrelia species available from GenBank. The most comparable sequences were of B. recurrentis (GenBank accession no. BQ346784), B. duttoni (GenBank accession no. BQ346788), B. crocidurae (GenBank accession no. AF247151), and B. persica (GenBank accession nos. AV530742 and EU914143), with 99%, 98%, 98%, and 97% resemblance, respectively. The sequence of B. microtti 16S-rRNA was less diverse from that of other Borrelia species, and it was found to be identical to that of B. duttoni (GenBank accession no. AF107366) and B. crocidurae (GenBank accession no. DQ057990). It also has 99% similarity with the isolates of B. recurrentis (GenBank accession no. BQ346813), B. hermsii (GenBank accession no. EF523250), and B. parkeri (GenBank accession no. AF307100).
Phylogenetic trees, generated from 16S-rRNA (523 bp), glpQ (668 bp), or combination of the two sequences (1188 bp) using neighbor joining method, demonstrated that the haplotype of B. microtti was related to B. duttoni and B. crocidurae, the African RF-associated Borrelia, and B. recurrentis, the agent of deadly louse-borne RF. On the other hand, B. persica haplotypes were associated to the new world RF-associated Borrelia such as B. parkeri and B. hermsii (Fig. 1).
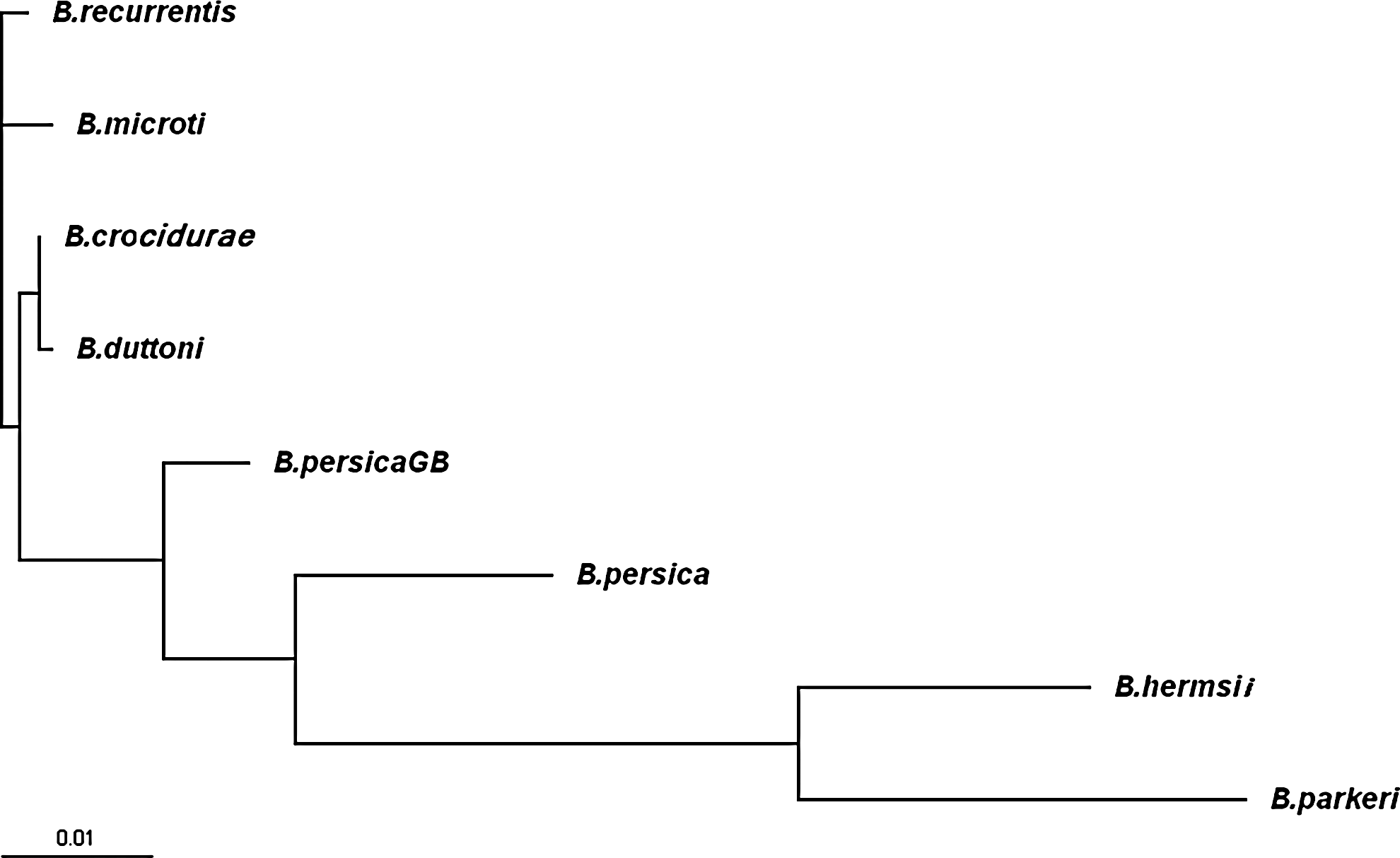
Phylogram inferred from sequence analysis of combined 1188 bp of glpQ (668 bp) and 16S-rRNA (520 bp) genes of different Borrelia species. Sequences of Borrelia recurrentis, Borrelia crocidurae, Borrelia duttoni, Borrelia hermsii, Borrelia parkeri, and Borrelia persicaGB have been retrieved from the GenBank database.
Development of species-specific PCR
Sequence analysis of the glpQ fragment allowed us to identify specific sequence elements for the design of appropriate primers. Species-specific primer for B. microtti (BMGlpQF: 5′-CCT TAG CGA AAG ATT TGA TCC T-3′) and B. persica (BPGlpQR: 5′-GCT GGG TCT TTG TTT TTT TGG-3′) worked successfully in combination with the universal reverse primer, 795R, and the forward primer, 128f, respectively. Using conventional PCR, the set of primers for B. persica amplified a 252-bp product, which was tested against the B. microtti strains. The set of primers for B. microtti amplified a 451-bp product, which was tested against the isolates of B. persica. Because no secondary bands were observed for both species, it is possible to use these primer sets to distinguish between the two species (Fig. 2). No amplification was observed when each of the species-specific primers was tested against the isolates of the other species.
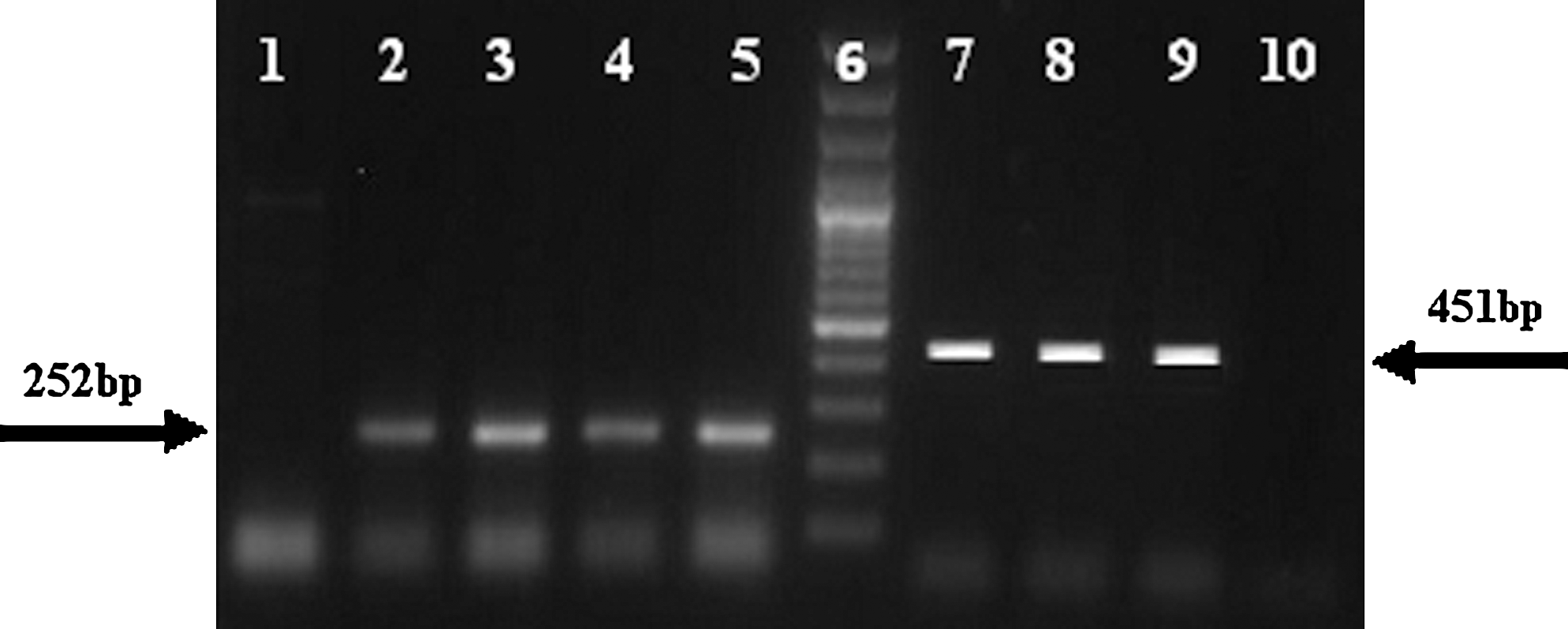
Species-specific polymerase chain reaction (PCR) of glpQ for Borrelia persica (252 bp) and Borrelia microtti (451 bp). Lanes: 1, B. microtti; 2–5, B. persica tested with B. persica-specific primer; 6, 100-bp ladder (Fermentas); 7–9, B. microtti; 10, B. persica tested by B. microtti-specific primer.
Sequence analysis of the 16S-rDNA lacks enough specific sequence elements for the design of appropriate primers. A few additional primers were designed and tested against strains of both species, but in all cases identical as well as secondary PCR products were observed (data not shown).
PCR-RFLP analysis of 16S-rRNA and glpQ
Sequence analysis of the glpQ fragment of the species revealed five specific restriction sites (DraI, TaqI, EcoRV, HinfI, and SspI) for B. persica versus B. microtti discrimination. All of these restriction enzymes gave two distinct patterns for each species (Fig. 3). The DraI digestion showed a profile of 293-, 226-, and 149-bp bands for B. persica, whereas for B. microtti, the 668-bp original PCR products remained intact. TaqI digestion revealed a profile of 261-, 194-, 156-, and 77-bp bands for B. persica, whereas it produced a profile of 484- and 184-bp bands for B. microtti. The PCR product of B. persica was digested by EcoRV into 534- and 134-bp fragments, whereas the PCR product for B. microtti was cut into 448-, 184-, and 86-bp fragments. The profile of HinfI for B. persica included 439- and 229-bp bands, whereas the B. microtti amplicon remained intact. Finally, the SspI digestion showed a profile of 309-, 185-, and 174-bp bands for B. microtti, whereas for B. persica it revealed a profile of 309-, 272-, and 86-bp bands.
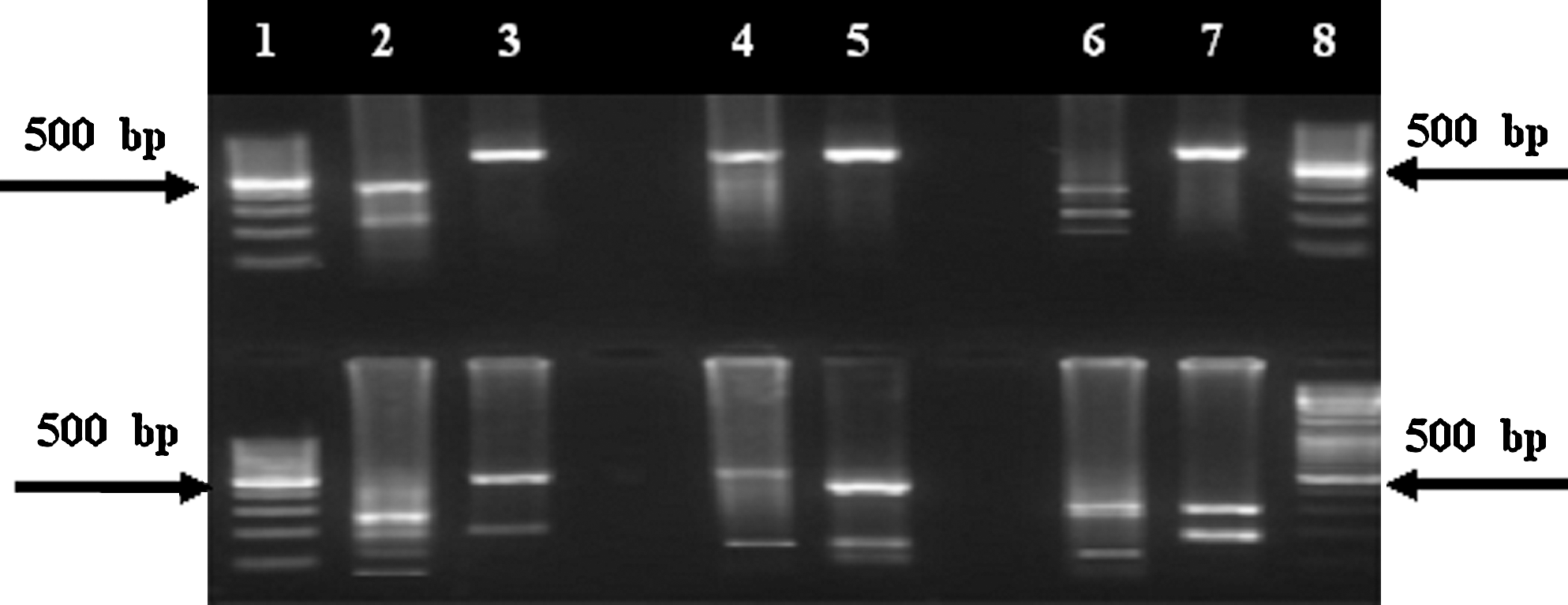
PCR–restriction fragment length polymorphism analysis of glpQ gene in Borrelia persica (lanes 2, 4, and 6) and Borrelia microtti (lanes 3, 5, and 7), using different restriction enzymes: HinfI (upper panel, lane 2: 439 and 229 bp; lane 3: 668 bp); controls: intact PCR (upper panel, lanes 4 and 5: 668 bp); DraI (upper panel, lane 6: 293, 226, and 149 bp; lane 7: 668 bp); TaqI (lower panel, lane 2: 261, 194, 156, and 77 bp; lane 3: 484 and 184 bp); EcoRV (lower panel, lane 4: 534 and 134 bp; lane 5: 448, 184, and 86 bp); and SspI (lower panel, lane 6: 309, 272, and 86 bp; lane 7: 309, 185, and 174 bp). Lanes 1 and 8 (upper and lower panels) are 100-bp ladder (Fermentas).
Sequence analysis of the 16S-rRNA fragment of the species revealed only an appropriate and specific restriction site “TaqI” for B. persica versus B. microtti discrimination. Restriction digestion of the 16S-rRNA PCR product with the enzyme TaqI gave two distinct patterns: B. persica showed a profile of 324- and 199-bp bands, whereas B. microtti showed a profile of 205-, 199-, and 119-bp bands (Fig. 4).
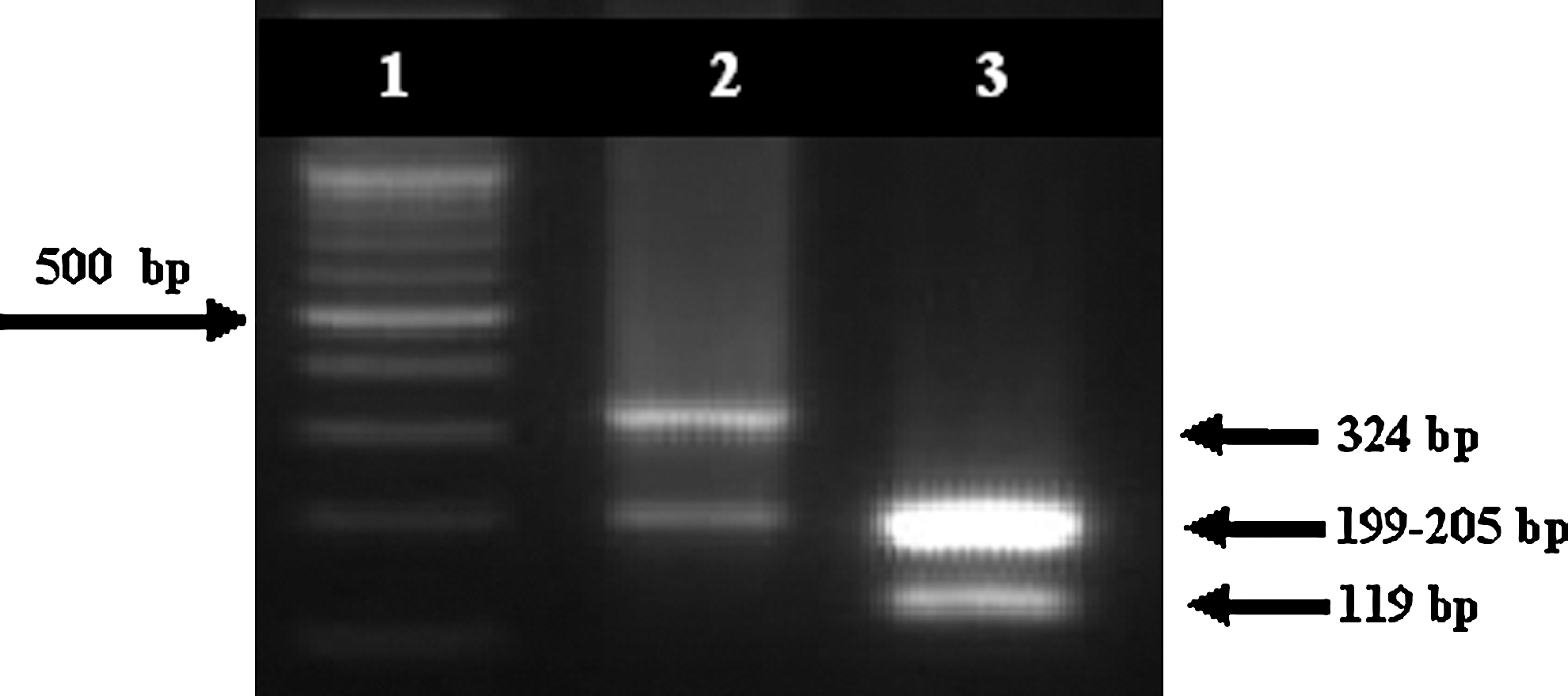
PCR–restriction fragment length polymorphism analysis of 16S-rRNA gene in Borrelia persica (lane 2) and Borrelia microtti (lane 3) with restriction enzyme TaqI. Lane 1: 100-bp ladder (Fermentas).
Discussion
Here, we describe the development of two reproducible discriminating PCR-based molecular typing methods (species-specific PCR and PCR-RFLP), which were able to characterize and distinguish strains of B. persica and B. microtti based on the amplification of the glpQ. The species-specific PCR is easier and faster than the PCR-RFLP method because the latter requires an extra postamplification step. Further, molecular typing of the species was developed using PCR-RFLP of 16S-rRNA. The high genetic variation of glpQ among the two Borrelia species studied here indicates that glpQ is a better candidate for discrimination between B. persica and B. microtti. The utility of glpQ as a useful marker for discrimination among other Borrelia species requires further investigation.
In tropical and subtropical areas, both TBRF and malaria occur, and because the main manifestation of both diseases is fever, TBRF is often obscured by malaria and incorrectly treated. On the other hand, RF is often atypical or causes mild or subclinical disease. Therefore, RF is ignored or often mistaken as malaria and hence incorrectly treated (Nordstrand et al. 2007, Larsson and Bergström 2008, Masoumi Asl et al. 2009). During fever peaks, diagnosis is simple because there is massive bacteraemia that could be easily detected, for example, Giemsa staining is used in malarial diagnosis. However, between the peaks and in milder cases, the bacterial concentration is very low and hard to find in a stained blood smear by microscopy. Protocols used for malarial diagnosis in which water is usually used to lyse erythrocytes also lyses spirochetes and are therefore useless for diagnosing RF. Performing phase-contrast or dark-field microscopy directly on 10-fold diluted blood is useful when spirochetemia is high. Because of the spirochetes' thin and transparent morphology, ordinary light microscopy is very difficult and of limited use. The inability of microscopic analysis to detect spirochetes gives rise to the need for tests with greater sensitivity.
Our literature review has shown that different techniques have been used for detection and diagnosis of Borrelia species so far. Detection is usually done during the primary attack by observation of spirochetemia on thin- or thick-blood smears with dark-field microscopy or with conventional microscopy after Giemsa, Wright, or Diff-Quick® staining (Barbour 1999, Johnson and Golightly 2000). Thick-blood smears provide 20 times the sensitivity of thin-blood smears. Quantitative buffy coat fluorescence analysis has also been described as a very sensitive and specific technique for detecting borreliae in blood (van Dam et al. 1999). Mouse inoculation has been used for a long time for isolation of RF Borrelia (Karimi et al. 1979), but RF borreliae can be cultured on axenic medium. Kelly's medium serves as the basis for Barbour Stoenner Kelly medium, which has been used for the cultivation of Lyme disease spirochetes as well as RF Borrelia recently (Cutler et al. 1999, van Dam et al. 1999). Culture and isolation of Borrelia takes about 21 days. The serological test, which is more often carried out for Lyme agents, takes less time but it is associated with a high number of false-positive reactions. To date, specific serological assays are not available for most of the known TBRFs, because of the lack of available antigens. If they were available, they could be useful if the blood smear is negative and PCR unavailable. However, serological methods cannot differentiate between active and past disease.
Cospeciation of each species of tick vector and its associated RF-causing Borrelia has been used conventionally for taxonomy of the Borrelia species. It is known that Borrelia–tick interactions are highly specific as each species of Borrelia is only transmitted by one or a few closely related species of ticks. O. tholozani is the main vector of B. persica, and its distribution overlaps the distribution area of clinical RF cases caused by B. persica. The tick is widely distributed throughout India and Kashmir, southern countries of the former USSR (Kazakhstan, Kyrgizia, Tajikistan, Turkmenistan, and Uzbekistan), and most of the Mediterranean and Middle East countries (Assous and Wilamowski 2009). Similarly, in Iran, Ornithodoros lahorensis was found to be infected with Borrelia spp. (Arshi et al. 2002), and O. erraticus is the vector of B. microtti, which causes a very mild RF (Assmar et al. 2002, Masoumi Asl et al. 2009). In Azerbaijan, Ornithodoros verrucosus is the vector of B. caucasica, which causes a very severe TBRF. In Iran and the countries of Central Asia of the former USSR, Ornithodoros tartakowskyi is the vector of B. latyschewii, which causes a very mild RF without apparent clinical relapses (Parola and Raoult 2001). Ornithodoros hermsi is capable of transmitting B. hermsii spirochete, whereas other species of ticks, such as Ornithodoros turicata and O. parkeri, are not. The other species of ticks are capable of transmitting other species of spirochetes (Schwan and Piesman 2002). The soft tick Ornithodoros sonrai is recognized as the only vector of B. crocidurae causing human RF in West Africa (Vial et al. 2006). The mechanisms responsible for this strict species specificity in transmission of one species of spirochete by only one species of tick are not known (Schwan and Piesman 2002).
All of these conventional methods aforementioned are usually quite slow, laborious, costly, and unable to efficiently identify species. By contrast, molecular methods are the most sensitive diagnostic tools used with increasing frequency and offer the possibility of species identification by employment of species-specific primers, PCR-RFLP, or sequencing, as shown in this work and previous studies (Fukunaga et al. 1996, Brahim et al. 2005, Halperin et al. 2006). Molecular typing can be used on different biological samples including ticks, buffy coat, cultures, and either fresh or frozen blood.
The molecular typing methods presented here are excellent diagnostic tools for differentiating B. persica versus B. microtti. Briefly, DNA is extracted from about 100 μL blood, analyzed by PCR using Borrelia-specific primers or PCR-RFLP, and then separated on an agarose gel. These methods are fairly quick and sensitive if the instrument and all reagents, including specific primers and enzymes, are accessible.
Footnotes
Acknowledgments
This work was supported by a grant from the Tehran University of Medical Sciences (TUMS) (grant no. 86-03-27-6268, to M.A.O.). The authors thank M. Abolhassani, A. Eskandari, and M. Hosseni from TUMS for technical assistance, and also Dr. Mehrdad Pedram (Department of Medical Genetics, TUMS) for his helpful comments and careful editing of the manuscript.
Disclosure Statement
No competing financial interests exist.