
Abstract
The specimens of 3552 questing adult Ixodes persulcatus and 1698 blood/tissue samples of small mammals collected in Ural, Siberia, and Far East of Russia were assayed for the presence of Anaplasma phagocytophilum by nested PCR based on the 16S rRNA gene. Totally, A. phagocytophilum was detected in 112 tick and 88 mammalian samples. The nucleotide sequences of the 16S rRNA gene and groESL operon (1244–1295 bp) were determined for A. phagocytophilum samples from 65 ticks and 25 small mammals. Six different 16S rRNA gene variants differing by 1–5 nucleotide substitutions were detected, and only one variant matched the sequences deposited in GenBank. Analysis of groESL sequences allowed the A. phagocytophilum samples to be divided into three groups; moreover, the samples from different groups also differed in the 16S rRNA gene sequences. The A. phagocytophilum sequences from group I were detected in 11 Myodes spp. samples from West Siberia and Far East and in 19 I. persulcatus samples from all examined regions; from group II, in 10 samples of Myodes spp. and common shrews (Sorex araneus) from Ural; and from group III, in four samples of Asian chipmunks (Tamias sibiricus) from West Siberia and Far East; and in 46 I. persulcatus samples from all examined regions. The nucleotide sequences of A. phagocytophilum groESL operon from groups I and II were strictly conserved and formed with A. phagocytophilum groESL sequence from a Swiss bank vole (Myodes glareolus) (GenBank accession no. AF192796), a separate cluster on the phylogenetic tree with a strong bootstrap support. The A. phagocytophilum groESL operon sequences from group III differed from one another by 1–4 nucleotides and formed a separate branch in the cluster generated by European A. phagocytophilum strains from roe deer (Capreolus capreolus) and Ixodes ricinus ticks.
Introduction
A transovarial transmission of A. phagocytophilum in Ixodes ticks has not been yet demonstrated, suggesting the need of reservoir hosts to maintain the life cycle of the pathogen. The most important reservoirs of A. phagocytophilum are white-footed mice (Peromyscus leucopus) and white-tailed deer (Odocoiles virginianus) in northern United States (Telford III et al. 1996, Tate et al. 2005) and dusky-footed wood rats (Neotoma fuscipes) and redwood chipmunks (Tamias ochrogenys) in western United States (Nieto and Foley 2009). In Europe, reservoir hosts of A. phagocytophilum are sheep, cattle, roe deer (Capreolus capreolus), red deer (Cervus elaphus), and different species of small rodents such as wood mice (Apodemus sylvaticum), voles (Myodes glareolus, Microtus agrestis, and Microtus oeconomus), and others (Ogden et al. 1998, Liz et al. 2000, 2002, Bown et al. 2003, Grzeszczuk et al. 2006).
The genetic diversity of A. phagocytophilum was studied by analyzing nucleotide sequences of both variable and conserved genes. Different variants of A. phagocytophilum 16S rRNA gene have been found; as a rule, they differ in a variable fragment located near the 5′ end of the gene (Massung et al. 2002, Cao et al. 2003); note that different genetic variants can circulate in the same biotope. In particular, six different A. phagocytophilum genetic variants were recorded in Norway (Stuen et al. 2006). The A. phagocytophilum genetic variants can considerably differ in the host tropism and pathogenic characteristics. Only two A. phagocytophilum genetic variants have been so far recorded in the blood of human cases (Chen et al. 1994, Chae et al. 2000). In addition to 16S rRNA gene, more variable genes (groESL operon, ank genes, major surface protein genes, citrate synthase genes, etc.) are used for characterizing and differentiating between the A. phagocytophilum strains circulating in various hosts (von Loewenich et al. 2003, de la Fuente et al. 2005, Cao et al. 2006).
I. persulcatus ticks are abundant in forest biotopes in Russia, from Baltic to the Far East. Unlike I. ricinus, immature I. persulcatus commonly infest birds and small mammals rather than large animals. Thus, small mammals are the most probable reservoirs for A. phagocytophilum. Currently, the A. phagocytophilum genetic variability in Russia is insufficiently studied despite A. phagocytophilum prevalence in I. persulcatus from different regions of Russia was studied in several researches (Eremeeva et al. 2006, Shpynov et al. 2006, Masuzawa et al. 2008, Nefedova et al. 2008).
The goal of this work was to study the genetic diversity of A. phagocytophilum in I. persulcatus and small mammals in Ural, Siberia, and Far East by analyzing the 16S rRNA gene and groESL operon.
Materials and Methods
Sampling
Adult I. persulcatus ticks were collected by flagging the vegetation in sampling sites from Ural (Sverdlovsk and Chelyabinsk regions), Siberia (Novosibirsk and Irkutsk regions), and Far East (Khabarovsk Territory) (Fig. 1). Wild small mammals were caught using live traps in sampling sites from Sverdlovsk, Novosibirsk regions, and Khabarovsk Territory. All experiments with animals were conducted in compliance with the Animal Welfare Act at the Institute of Systematics and Ecology of Animals, Siberian Branch, Russian Academy of Sciences, according to the guidelines for experiments with laboratory animals (Supplement to the Order of the Russian Ministry of Health No. 755 of August 12, 1977). The trapped animals were anesthetized with diethyl ether. Blood and/or spleen samples were taken from animals trapped in Sverdlovsk and Novosibirsk regions. Blood aliquots (200–300 μL) were sampled into the tubes with 50 mM EDTA. Spleen and liver samples were taken from animals trapped in Khabarovsk Territory. Parts of the spleen and liver specimens from each animal (∼50 mg) were pooled for DNA extraction. Ticks and tissue/blood samples were stored at −20°C until use.
Sampling sites of Ixodes persulcatus ticks and small mammals: (1) Sverdlovsk region, North Ural; (2) Sverdlovsk region, Middle Ural; (3) Chelyabinsk region, South Ural; (4) Novosibirsk region, West Siberia; (5) Irkutsk region, East Siberia; and (6) Khabarovsk Territory, Far East.
Sampling sites and sample types are given in the following list: (1) Sverdlovsk region, North Ural (60°N, 59°E); 496 ticks collected in May–June 2004–2006 and 2009–2010 and blood and/or spleen samples of 199 small mammals—143 voles (Myodes spp.), 53 shrews (Sorex spp.), and 3 field voles (Mi. agrestis), captured in June–July 2004 and 2005 in the Denezkin Kamen’ Reserve. (2) Sverdlovsk region, Middle Ural (57°N, 60°E); 145 ticks collected in May–June 2007, 2008 and blood and/or spleen samples of 50 small mammals—31 Myodes spp., 16 common shrews (Sorex araneus), and 3 striped field mice (Apodemus agrarius), captured in June–July 2008 in the Visim Reserve. (3) Chelyabinsk region, South Ural (55°N, 60°E); 79 ticks collected in May 2004. (4) Novosibirsk region, West Siberia (55°N, 83°E); 1373 ticks collected in May–June 2003, 2004, and 2006–2010; blood and/or spleen samples of 307 small mammals—143 Myodes spp., 73 S. araneus, 11 Ap. agrarius, 38 voles (Microtus spp.), 36 northern birch mice (Sicista betulina), and 6 Siberian chipmunks (T. sibiricus), captured in May–September 2003, 2006, 2007, and 2010. (5) Irkutsk region, East Siberia (52–58°N, 101–114°E); 928 ticks collected in May–June 2006–2009 at different sites of the region. (6) Khabarovsk Territory, Far East (48°N, 135°E); 730 ticks collected in April–June 2006–2009; spleen and liver samples of 1142 small mammals—709 Myodes spp., 56 S. araneus, 359 Korean field mice (Apodemus peninsulae), and 18 T. sibiricus, captured in February–November 2006–2010.
DNA extraction
Total DNA was extracted from crushed ticks or blood/tissue samples as previously described (Rar et al. 2010).
PCR assay
PCR assay was conducted as previously described (Rar et al. 2010). PCR mixtures contained 67 mM Tris-HCl (pH 8.9), 16.6 mM (NH4)2SO4, 2 mM MgCl2, 0.01% Tween 20, 200 μM of each dNTP, 5% glycerol, 0.5 μM primers, 2 U of Taq DNA polymerase (“Biosan,” Novosibirsk, Russia), and 2 μL of DNA for primary reactions or 2 μL of the primary PCR products for nested reactions. For screening analysis, A. phagocytophilum DNA was detected by nested PCR using primers targeted to the 16S rRNA gene. Primers Ehr1 and Ehr2 were used for primary reactions and HGE1 and HGE2 for nested reactions (Table 1). The length of nested PCR fragments was 494 bp. For sequencing analysis, the 16S rRNA gene fragments of about 1350 bp were amplified using primers Ehr1 and Ehr6 for the primary reactions and Ehr7 and Ehr8 for the nested reactions. The groESL operon fragments of about 1320 bp were amplified with primers HS1-f and HS6-r for the primary reactions and HS3-f and HSVR for nested reactions (Table 1).
Sequencing of PCR products
Nucleotide sequences of PCR products purified on GFX Columns (Amersham Biosciences, Piscataway, NJ) were determined at the DNA Sequencing Center of the Siberian Branch, Russian Academy of Sciences, Novosibirsk (
Nucleotide sequence accession numbers
The partial 16S rRNA gene and groESL operon nucleotide sequences of A. phagocytophilum were deposited in GenBank under accession numbers HM366567–HM366590 and HQ630614–HQ630625.
Results
A. phagocytophilum prevalence
Totally, samples of 3751 questing adult I. persulcatus ticks and blood/tissue samples of 1698 small mammals from different sites in Ural, Siberia, and the Far East were examined for the presence of A. phagocytophilum using nested PCR based on the 16S rRNA gene. A. phagocytophilum DNA was found in 112 samples of ticks and in 88 samples of small mammals. In different regions, the prevalence rate of A. phagocytophilum in ticks varied from 0.2% to 6.3% and, in small mammals, from 2.0% to 16.6% (Tables 2 and 3). The infected individuals represented the majority of the studied species, namely, red-backed vole (Myodes rutilus), red gray-backed vole (M. rufocanus), bank vole (M. glareolus), common shrew (S. araneus), East-European field vole (Microtus rossiaemeridionalis), Korean field mouse (Ap. peninsulae), and Siberian chipmunk (T. sibiricus) (Table 3).
A. phagocytophilum was detected using nested PCR based on the 16S rRNA gene.
A. phagocytophilum was detected using nested PCR based on the 16S rRNA gene.
A. phagocytophilum 16S rRNA gene variability
The nucleotide sequences of A. phagocytophilum 16S rRNA gene and groESL operon were determined in 65 I. persulcatus and 25 samples of small mammals (Myodes spp., S. araneus, and T. sibiricus) from different regions. Totally, six variants of 16S rRNA gene sequences differing in one to five nucleotide substitutions were detected; note that only the first variant matched the sequences previously deposited in GenBank (Table 4).
Positions of variable nucleotides are given according to the A. phagocytophilum 16S rRNA gene sequence (GenBank accession no. AF093788).
The reference A. phagocytophilum 16S rRNA gene sequences.
Samples from Ixodes persulcatus.
Samples from Myodes spp.
Samples from T. sibiricus.
The nucleotide sequences of A. phagocytophilum 16S rRNA gene determined in 17 I. persulcatus from different regions and 11 Myodes spp. samples from the Novosibirsk region and Khabarovsk Territory (variant 1) were identical to one another and to the sequence of a widespread A. phagocytophilum genetic variant with a zoonotic potential (CAHU-HGE1; GenBank accession no. AF093788) (Chae et al. 2000). The 16S rRNA gene sequences determined in two ticks from the Irkutsk region (variant 2) and 10 Myodes spp. and S. araneus samples from North and Middle Ural (variant 3) differed from the corresponding sequence of variant 1 by a single-nucleotide substitution in the conserved region of the gene. A. phagocytophilum 16S rRNA gene sequences determined in 43 I. persulcatus from Novosibirsk region, Irkutsk region, and Khabarovsk Territory and four chipmunk samples from the Novosibirsk region and Khabarovsk Territory (variant 4) were most similar (differing by one nucleotide in the conserved region of the gene) to the sequence of a rare A. phagocytophilum genetic variant (GenBank accession no. DQ342324), detected earlier in I. persulcatus and small mammals only in China (Cao et al. 2006). In addition, A. phagocytophilum 16S rRNA gene sequences determined in one tick from the Irkutsk region (variant 5) and two ticks from the Sverdlovsk region (variant 6) differed from the corresponding sequence of variant 4 by a single-nucleotide substitution in the conserved region of the gene (Tables 4 and 5).
I–III, A. phagocytophilum groups based on groESL operon sequences; IIIa–IIIh represent different sequences of group III.
1–6: A. phagocytophilum variants based on the 16S rRNA gene sequences.
Samples from I. persulcatus.
Samples from Myodes spp.
Samples from T. sibiricus.
A. phagocytophilum groESL operon variability
According to analysis of groESL operon sequences, all the detected A. phagocytophilum samples fell into three groups; note that the samples belonging to different groups also differed in their 16S rRNA gene sequences (Table 5). The homology between the groESL operon sequences was 94.8%–98.3% in different groups and 99.7%–100% within groups (Fig. 2).
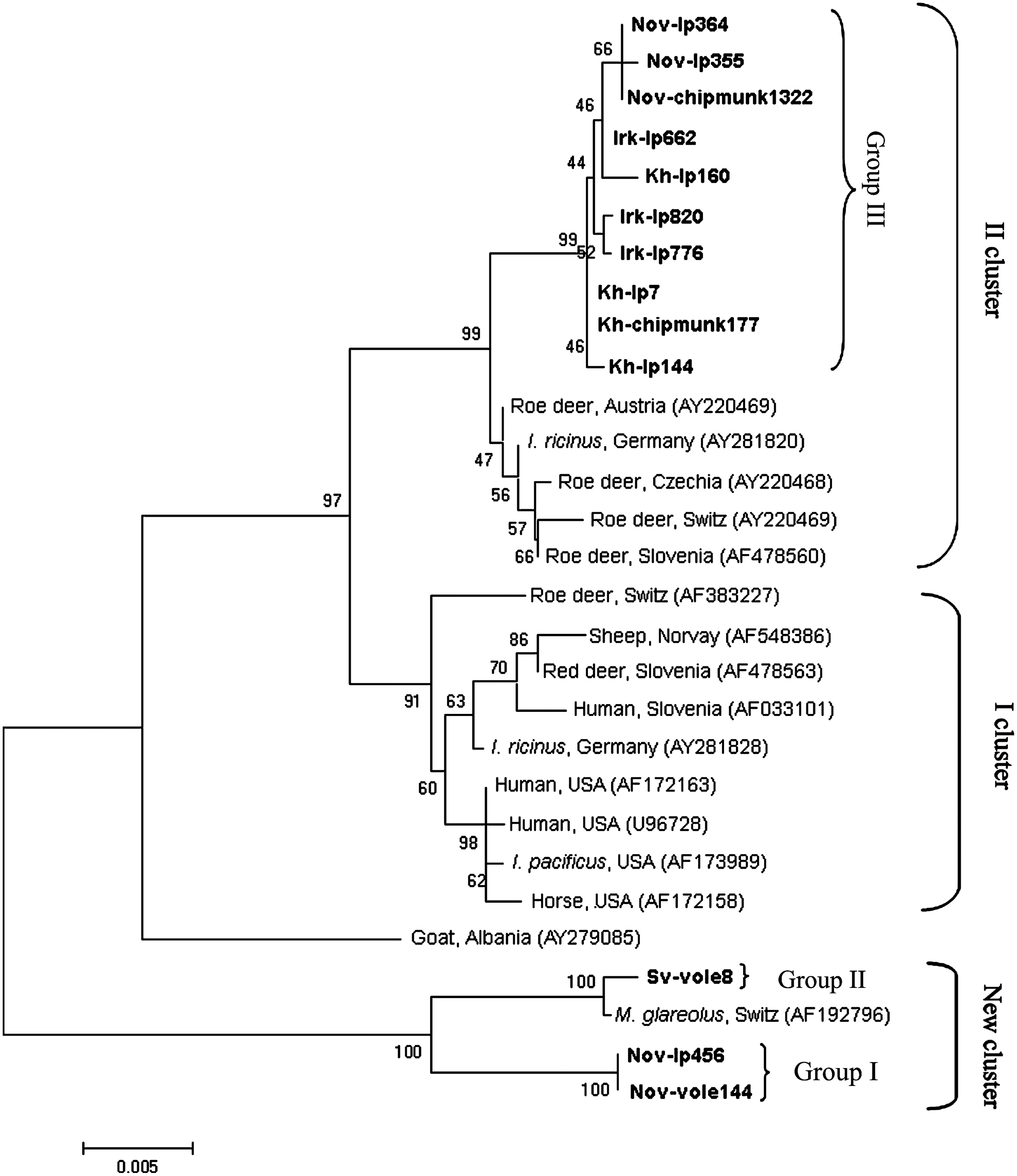
The phylogenetic tree constructed by the neighbor-joining method using the 1245-bp sequences of Anaplasma phagocytophilum groESL operon. The scale bar indicates an evolutionary distance of 0.005 nucleotides per position in the sequence. The sequences of A. phagocytophilum from different sources are represented on the phylogenetic tree; GenBank accession numbers are given in parentheses. Groups I–III comprise sequences determined in this study. The specimens from I. persulcatus (Nov-Ip456, Nov-Ip355, Nov-Ip364, Irk-Ip662, Irk-Ip776, Irk-Ip820, Kh-Ip7, Kh-Ip144, and Kh-Ip160), Myodes spp. (Nov-vole144 and Sv-vole8), and Siberian chipmunks (Nov-chipmunk1322, and Kh-chipmunk177) in this study, representing different sequence variants, are in boldface.
A. phagocytophilum groESL operon sequences belonging to group I were detected in 19 I. persulcatus from all studied regions and in 11 Myodes spp. samples from the Novosibirsk region and Khabarovsk Territory. All the determined sequences were identical to each other (typical sequences in the dendrogram, Nov-Ip456 and Nov-vole144) but differed from the groESL operon sequences available in GenBank. The highest similarity (98.2%) was recorded for the A. phagocytophilum groESL operon sequence of a Swiss bank vole (GenBank accession no. AF192796) (Liz et al. 2000); the similarity to other groESL sequences did not exceed 95.5%.
A. phagocytophilum groESL operon sequences from group II were detected only in the samples of voles and common shrews from the Sverdlovsk region (nine samples from North Ural and one from Middle Ural). All the determined groESL sequences were also identical to one another (a typical sequence in the dendrogram, Sv-vole8) and differed by only three of the overall 1246 nucleotides (99.8% homology) from the A. phagocytophilum groESL sequence of a Swiss bank vole (GenBank accession no. AF192796) (Liz et al. 2000); the similarity to other groESL sequences did not exceed 95.4%.
A. phagocytophilum groESL operon sequences from group III were detected in 46 I. persulcatus from all the studied regions and in four chipmunk samples from the Novosibirsk region and Khabarovsk Territory. Group III was heterogeneous and contained eight different A. phagocytophilum groESL operon sequences (denoted IIIa–IIIh in Table 5) differing by 1–4 nucleotides and most similar (99.2%–99.4% homology) to a number of A. phagocytophilum groESL operon sequences from I. ricinus and roe deer (GenBank accession nos. AY220469 and AY281820) (Petrovec et al. 2002, von Loewenich et al. 2003). The dendrogram in Figure 2 shows typical samples of group III: Nov-Ip355, Nov-Ip364, Irk-Ip662, Irk-Ip776, Irk-Ip820, Kh-Ip7, Kh-Ip144, Kh-Ip160, Nov-chipmunk1322, and Kh-chipmunk177. The 16S rRNA gene sequences of A. phagocytophilum samples from group III differed from the known sequences of European isolates and were most similar to the 16S rRNA gene sequences of the A. phagocytophilum isolates from China.
Phylogenetic analysis showed that A. phagocytophilum groESL operon sequences from groups I and II formed with the sequence from a Swiss bank vole a separate cluster on the phylogenetic tree with a strong bootstrap support, whereas A. phagocytophilum groESL operon sequences from group III formed a separate branch in the cluster generated by European A. phagocytophilum strains from roe deer and I. ricinus (Fig. 2). The prevalence of different A. phagocytophilum variants in ticks and small mammals from different regions is listed in Table 5. In all studied regions, A. phagocytophilum groESL operon sequences from groups I and III were determined in ticks; however, different sequence variants of group III prevailed in different regions. Only A. phagocytophilum of group I was determined in voles in Novosibirsk region and Khabarovsk Territory, and only A. phagocytophilum of group II was determined in voles and shrews in Sverdlovsk region. Only A. phagocytophilum of group III was determined in chipmunks and the same sequence variants of group III were found in ticks and chipmunks in Novosibirsk region as well as in Khabarovsk Territory.
Discussion
In this study, A. phagocytophilum was found in ticks and small mammals in all examined regions of Ural, Siberia, and Far East. It has been demonstrated that different hosts vary in susceptibility to local A. phagocytophilum strains (Foley et al. 2008) and thus the study of A. phagocytophilum genetic variability is important for understanding the risk of A. phagocytophilum infections in human and domestic animals.
The groESL heat shock operon is widely used for differentiating A. phagocytophilum isolates. So far, about 40 different variants of groESL operon from Europe and the United States have been detected; the majority of them fall into two clusters (Petrovec et al. 2002, von Loewenich et al. 2003, Alberti et al. 2005, Rymaszewska 2008, Katargina et al. 2011). The first cluster contains the A. phagocytophilum sequences from a wide range of vertebrate hosts, including deer, roe deer, sheep, horses, human cases, and I. ricinus ticks. The second cluster comprises only European A. phagocytophilum strains from roe deer and I. ricinus. Note that at least two A. phagocytophilum groESL operon sequences cannot be ascribed to either cluster, namely the A. phagocytophilum sequences isolated from a goat in Albania (GenBank accession no. AY279085) and a Swiss bank vole (GenBank accession no. AF192796) (Liz et al. 2000, Petrovec et al. 2003).
In this study, it was determined that A. phagocytophilum groESL operon sequences from Ural, Siberia, and Far East were attributed to three distinct groups. A. phagocytophilum groESL operon sequences of group III, which were determined in most of I. persulcatus and in chipmunks samples, belong to the second cluster associated with roe deer. Note that none of the determined groESL operon sequences can be attributed to the first cluster associated with human and domestic animal infections (Fig. 2). On the other hand, A. phagocytophilum groESL operon sequences from groups I and II, which were found in voles, shrews, and some of I. persulcatus samples, do not belong to any of previously described clusters. These sequences together with the only one known A. phagocytophilum groESL operon sequence from small mammals in Europe, that of a Swiss bank vole (GenBank accession no. AF192796) (Liz et al. 2000), form a new cluster on the phylogenetic tree.
Interestingly, the A. phagocytophilum groESL operon sequences from the new cluster associated with voles and shrews are highly conserved, unlike the corresponding sequences of the other two clusters. Only three different groESL operon sequences belonging to this cluster have been found: the first sequence was detected in five Swiss bank voles (Liz et al. 2000); the second, in 10 voles and shrews from Ural; and the third, in 11 voles from Siberia and the Far East and in 19 I. persulcatus ticks.
Note that the A. phagocytophilum genetic variants detected in small mammals from Switzerland and Ural are closely related to each other (99.8% homology based on groESL operon sequences) and the corresponding groESL operon sequences have not yet been detected in Europe in I. ricinus ticks (Liz et al. 2007; von Loewenich et al. 2003; Katargina et al. 2011) or in I. persulcatus in this study. It was recently demonstrated on the basis of msp4 sequence analysis that field voles in northern England also harbor the unique A. phagocytophilum genotype, which markedly differed from genotypes found in I. ricinus and roe deer (Bown et al. 2009).
In this study, A. phagocytophilum was detected in 33 of 199 (16.6%) examined small mammals from North Ural and only in one of the 496 adult I. persulcatus collected in the same area of North Ural during the 5-year period (unfortunately, the genetic variant of this sample was not detected). However, A. phagocytophilum was found in two I. persulcatus larvae fed on an infected common shrew and bank vole (data not shown). Thus, we can suppose that I. persulcatus is an inefficient vector for A. phagocytophilum genetic variant associated with small mammals from North Ural and that other ixodid ticks can be responsible for the transmission of A. phagocytophilum to small mammals. In addition to I. persulcatus, the nidicolous ticks I. trianguliceps are abundant in the studied North Ural region (Livanova and Livanov 2010). The vector competence of I. trianguliceps for A. phagocytophilum has been well established (Ogden et al. 1998, Bown et al. 2003), and it was recently suggested that in northern England I. trianguliceps but not I. ricinus can transmit A. phagocytophilum to voles (Bown et al. 2009). Thus, we suppose that I. trianguliceps is the most probable vector for A. phagocytophilum of group II distributed in northern Ural.
Thus, we have found three distinct A. phagocytophilum lineages in the Asian part of Russia; one of them is associated with the voles of West Siberia and Far East, the second with Ural voles and shrews, and the third with chipmunks. Any competent vector for the A. phagocytophilum genetic variant circulating in small mammals in Ural has not been detected. Further studies of other variable genes will be useful for a deeper insight into the new A. phagocytophilum genetic variants discovered in this work.
Footnotes
Acknowledgment
This work was supported by the Siberian Branch of the Russian Academy of Sciences (integration projects nos. 6 and 63).
Disclosure Statement
No competing financial interests exist.