
Abstract
Rickettsia parkeri, a spotted fever group (SFG) rickettsia recently found to be pathogenic to humans, causes an eschar-associated febrile illness. The R. parkeri rickettsiosis, Tidewater spotted fever, has been misdiagnosed as Rocky Mountain spotted fever due to serologic cross reactivity and the lack of specific diagnostic methods. Candidatus Rickettsia andeanae, also a SFG rickettsia, is a recently described agent of unknown pathogenicity originally identified in ticks collected from domestic animals during a fever outbreak investigation in northern Peru. Among 37 Amblyomma maculatum (collected from humans (n=35) and questing (n=2)) obtained from the southern United States during 2000–2009, nine and four A. maculatum nucleic acid preparations were found positive for R. parkeri and Candidatus R. andeanae, respectively, by newly developed genus- and species-specific quantitative real-time polymerase chain reaction assays. In addition Rickettsia felis was found in two A. maculatum nucleic acid preparations.
Introduction
Antibodies to spotted fever group (SFG) rickettsiae, sometimes at high prevalences (2.5 to 28.7%), have been detected in many groups of humans investigated for evidence of previous infection with Rickettsia rickettsii, the causative agent of Rocky Mountain spotted fever (RMSF) in the United States (Wilfert et al. 1985, Dasch et al. 1993, Richards et al. 1993, Yevich et al. 1995, McCall et al. 2001, Marshall et al. 2003, Graf et al. 2008). However, it is now thought that these high prevalences of antibodies to SFG rickettsiae are due not only to infection with R. rickettsii, but also to infection with other SFG rickettsial agents (Taylor et al. 1985, Dasch et al. 1993, Marshall et al. 2003, Parola et al. 2005, Graf et al. 2008, Paddock et al. 2008, Stromdahl et al. 2011). With the recent determination that R. parkeri can cause an SFG rickettsiosis (Paddock et al. 2004) and that this rickettsiosis can be misdiagnosed as RMSF due to serologic cross reactivity (Paddock et al. 2008, Parola et al. 2009), a specific assay to detect R. parkeri is warranted.
Candidatus Rickettsia andeanae, an incompletely characterized rickettsia, was initially described during a fever outbreak investigation in northern Peru that was determined to be partially associated with an SFG rickettsiosis (Blair et al. 2004b). Characterization of Candidatus R. andeanae by molecular techniques, polymerase chain reaction (PCR), and multilocus sequence typing (MLST) showed it to be a unique SFG rickettsia based on sequences of PCR segments amplified from five well-known genes (17 kDa antigen gene, gltA, ompB, ompA, and sca4) (Blair et al. 2004a, Jiang et al. 2005).
Candidatus R. andeanae was originally detected in one A. maculatum and one Ixodes boliviensis tick removed from two different domestic horses from two different villages in the Department of Piura in northwestern Peru (Blair et al. 2004a). A subsequent investigation of rickettsiae in arthropods collected from domestic animals from the Department of Piura was conducted in 2007. Candidatus R. andeanae was detected in 26 A. maculatum and two Rhipicephalis sanguineus ticks collected from domestic animals (horses, donkeys, cows, and dogs) in all three districts evaluated. In addition, it was found to be sympatric in one district with R. parkeri (found in seven A. maculatum) and Rickettsia felis (found in two Ctenocephalides canis) (Florin et al. 2011). To date, Candidatus R. andeanae has yet to be cultured from ticks, humans, or other animals, and many characteristics of this rickettsia such as pathogenicity and distribution remain unknown.
This article describes the development and validation of quantitative real-time PCR (qPCR) assays to detect R. parkeri and Candidatus R. andeanae, and the molecular evidence of rickettsiae in A. maculatum submitted to the DoD Human Tick Test Kit Program of the U.S. Army Center for Health Promotion and Preventive Medicine (USACHPPM) (Stromdahl et al. 2001) during 2000–2009. The newly developed qPCR assays identified both R. parkeri and Candidatus R. andeanae in Gulf Coast ticks from three and two southern states, respectively.
Materials and Methods
Primers and probe design
For the development of the Rpark qPCR assay, the sequence of R. parkeri strain Mac20 ompB was aligned with 22 different Rickettsia species by using Vector NTI 11.0 (Invitrogen, Carlsbad, CA). A 24 bp sequence unique for R. parkeri was chosen to design the molecular beacon probe (Rpa188probe: 6-FAM-CGCGAAATTAATACCCTTATGAGCAGCAGTCGCG-BHQ-1). The primers (Rpa129F: CAAATGTTGCAGTTCCTCTAAATG and Rpa224R: AAAACAAACCGTTAAAACTACCG) for the assay produce a 96 bp product.
For the development of the Rande qPCR assay, a species-specific sequence of Candidatus R. andeanae strain T163 ompB was used as the molecular beacon probe (Rand1003FAM: 6FAM-CGCGATGTAGGCGGACAGGTAACTTTTGATCGCG-BHQ-1). The primers (Rand957F: CGCTGGACAAGTTTATGCTCAAG and Rand1062R: GGCAGTAGTACCGTCTGTACCAAC) for the assay amplify a 109 bp segment. Beacon Designer 4.0 (Biosoft International, Palo Alto, CA) was used to evaluate the suitability of the selected primers as well as the probes for both assays.
For the development of the Rick17b qPCR assay, a modification of the Rickettsia genus-specific Rick17 qPCR assay (Jiang et al. 2004) was accomplished with a new primer set (R17K128F2: GGGCGGTATGAAYAAACAAG and R17K238R: CCTACACCTACTCCVACAAG). The TaqMan probe R17K202TaqP (6FAM-CCGAATTGAGAACCAAGTAATGC-TAMRA) for Rick17b targets the same 23 bp sequence in the Rick17 qPCR assay. When run side by side, these two assays against 11 rickettsial DNA in the panel, the cycle threshold value from Rick17b was 1.6 to 3 less than Rick17 (data not show).
Optimal conditions for Rpark, Rande, and Rick17b qPCR assays
Rpark and Rande qPCR assays were optimized in a SmartCycler II (Cepheid, Sunnyvale, CA) thermocycler system by using OmniMix HS beads (Cepheid). Each 25 μL reaction contained 1 μL of template DNA, 0.7 μM of forward and reverse primers, 0.4 μM of probe, and 8 mM of MgCl2 for the Rpark and 0.5 μM of forward and reverse primers, 0.4 μM of probe, and 6 mM of MgCl2 for the Rande assays. The cycler parameters included initial denaturation of 2 min at 94°C and 45–50 cycles of denaturation for 5 s at 94°C and annealing/elongation for 30 s at 60°C for Rpark and 62°C for Rande qPCR assays. The Rick17b qPCR assay (detects the Rickettsia genus-specific 17 kDa antigen gene) was optimized with Platinum Quantitative PCR SuperMix-UDG (Invitrogen) using primers at 0.5 μM, probe at 0.4 μM, and MgCl2 at 5 mM. The 25 μL reactions contained 1 μL of template DNA and were run on a SmartCycler II (Cepheid) thermocycler system with 2 min at 50°C, 2 min at 95°C, and 45–50 cycles of denaturation for 15 s at 95°C and annealing/elongation for 30 s at 60°C. For the optimization of the assays, the concentrations of the primers were varied from 0.2 to 0.8 μM with steps of 0.1 μM, the probe was varied from 0.3 to 0.7 μM with steps of 0.1 μM, and the MgCl2 from 4 to 8 mM (10 mM for Rpark assay) with steps of 1 mM. The annealing temperatures varied from 58°C to 62°C with steps of 1°C.
Determination of Rpark, Rande, and Rick17b qPCR assays analytical specificity
To determine the ability of the Rpark, Rande, and Rick17b qPCR assays to distinguish R. parkeri, Candidatus R. andeanae, and Rickettsia DNA, respectively, from rickettsial (n=20) and nonrickettsial (n=12) DNA a panel of bacterial nucleic acid preparations representing near and far neighbors (Table 1) was used in a similar manner to that previously described (Jiang et al. 2004).
Determination of the limit of detection for Rpark, Rande, and Rick17b qPCR assays
To determine the limit of detection (LOD) of the Rpark, Rande, and Rick17b qPCR assays, a plasmid DNA for each assay was constructed by ligating PCR amplicons consisting of species- or genus- specific target sequences into pCR-XL-TOPO vectors by using TOPO XL PCR Cloning Kit (Invitrogen). The concentrations of the three plasmid preparations (copy numbers/μL) were calculated following their measured absorbance at 260 nm with a BioPhotometer (Eppendorf, Hauppauge, NY). Serial dilutions (log 10 dilutions from 108 to 103, then half log dilutions down to 100) of these plasmid preparations were made to produce standard curves and determine the LOD for each assay. At least two sets of the plasmid dilutions were made (more dilution series were tested at the low range of less than 102 copies/μL) and used to determine the LOD. In addition, human genomic DNA (Roche Applied Sciences, Indianapolis, IN) was added to the serial dilutions to a final concentration of 25 μg/mL (Loftis et al. 2003) to evaluate the performance of the assays in a background of human DNA.
Validation of Rpark and Rande qPCR assays with nucleic acid extracted from human and tick samples
Known infected and uninfected ticks (Stromdahl personal communication) and human clinical specimens (Whitman et al. 2007) were used to validate the effectiveness of the Rpark qPCR assay in detecting R. parkeri among clinical and environmental samples. Fifty-four A. maculatum in 37 nucleic acid preparations (30 individual ticks removed from humans, 5 pools of ticks removed from humans, and 2 questing ticks) submitted to the DoD Human Tick Test Kit Program of the USACHPPM from 11 southern states during 2000–2009 were sent to the Naval Medical Research Center (NMRC) blinded as to their PCR/RFLP results. Nucleic acid preparations from serum, blood, and tissue biopsies of the eschar and a rash papule of a human case of Tidewater spotted fever (Whitman et al. 2007) were also assessed.
To validate the Rande qPCR assay, nucleic acid preparations from two ticks identified as having Candidatus R. andeanae by PCR/MLST, two ticks determined as having SFG rickettsiae, and two ticks negative for rickettsiae (Blair et al. 2004a) were tested with the Rande qPCR assay.
In addition, Rande and Rick17b qPCR assays were used to further investigate the A. maculatum nucleic acid preparations initially tested negative by Rpark for the presence of Candidatus R. andeanae and other rickettsiae.
PCR and DNA sequencing
To confirm the positive results from Rande and Rick17b qPCR assays, standard PCR and half-nested PCR were performed to amplify a segment of sca4 by using Platinum PCR SuperMix High Fidelity (Invitrogen) on a T-Gradient Thermocycler (Biometra, Goettingen, Germany). The PCR amplicons were purified and sequenced by using BigDye Terminator v 3.1 Ready Reaction Cycle Sequencing Kits (Applied Biosystems, Foster City, CA) on an automated ABI Prism 3130xl genetic analyzer (Applied Biosystems) similar to that previously described (Jiang et al. 2005). Primers used for PCR amplification and sequencing are listed in Table 2. The sequences were assembled by using Vector NTI advance 11 software (Invitrogen), and BLAST searches were performed on NCBI website (
PCR amplicon size from primer set D1F and RrD1826R.
Half-nested PCR amplicon size from primer set D1F and D928R.
Half-nested PCR amplicon size from primer set RrD749F and RrD1826R.
PCR, polymerase chain reaction.
Results
Optimizations of the Rpark and Rande qPCR assays were performed by using nucleic acid preparations from R. parkeri C and Candidatus R. andeanae T163, respectively. To determine the specificity of these assays nucleic acid preparations from 20 different species of Rickettsia, 12 species of nonrickettsial bacteria, and human mononuclear cells were assessed. Only the R. parkeri and Candidatus R. andeanae nucleic acid preparations were positive for the Rpark and Rande qPCR assays, respectively. In addition, all rickettsial DNA preparations were detected by the Rick17b Rickettsia-specific qPCR assay. All other near and far neighbor preparations assessed were negative (Table 1) as was the human mononuclear cell nucleic acid preparation (data not shown).
Serial dilutions of target sequence containing plasmid preparations from 108 to 100 were made, from which individual standard curves were created to determine the LOD of each assay and to quantify unknown samples. The LOD for the Rpark qPCR assay was found to be consistent at the level of 3 copies of target gene/reaction (7/7), though the assay was capable of detecting a single copy/reaction 67% (6/9) of the time. The amplification efficiency of Rpark assay was 95% with r 2 value of 0.998. The LOD for the Rande and Rick17b qPCR assays were also found to be three copies/reaction. The amplification efficiency and the r 2 values of the Rande and Rick17b assays were 95% and 0.999, and 104% and 0.996, respectively. The efficiency of the qPCR assays was not affected by the addition of human DNA at a final concentration of 25 μg/mL to the plasmid controls, though a decrease in total fluorescence was noted in the 102 to 100 copies/reaction of the samples when compared with the plasmid controls without human DNA.
To evaluate the effectiveness of the Rpark qPCR assay and to determine its ability to detect R. parkeri in the presence of host genome nucleic acid, it was assessed with clinical and environmental samples known to contain R. parkeri DNA. DNA extracted from blood, serum, and skin-biopsy specimens of the eschar and a shoulder papule from a Tidewater spotted fever patient (Whitman et al. 2007) were tested by Rpark qPCR. DNA from the eschar and papule showed a positive reaction, but negative reactions were obtained for the DNA preparations from blood and serum samples. These results were identical to those previously obtained by PCR/MLST (Whitman et al. 2007). To further validate the Rpark qPCR assay, the 37 A. maculatum nucleic acid preparations (9 R. parkeri-positive and 28 R. parkeri-negative) previously tested at the USACHPPM by PCR/RFLP analysis with primer pair Rr190.70p and 190.602n and pstI (Stromdahl et al. 2008) were used. These samples were assessed with the Rpark qPCR assay by the NMRC laboratory and blinded to the results obtained at USACHPPM. The results of the Rpark qPCR assay were identical to those previously determined by PCR/RFLP analysis (Table 3) and show the distribution of R. parkeri infected A. maculatum ticks found on people reporting to DoD health care centers in Florida (n=2; Hurlburt Field in the panhandle of western Florida), Kentucky (n=5; Fort Campbell in southern KY/northern TN), and Virginia (n=1; Fort Eustis in the Tidewater area of VA), and questing (sample #2) in Kentucky (n=1; Fort Campbell, KY) (Fig. 1). The positive samples from Fort Campbell, KY, were collected at six different times from May to August during a 7-year period (Jul 2003, Jun 2005, May 2006 (questing), Aug 2007, August 2008, and August 2009). Detection of R. parkeri nucleic acid in patient samples and in A. maculatum ticks by Rpark qPCR assay shows the strength of this assay when used with clinical and environmental samples.
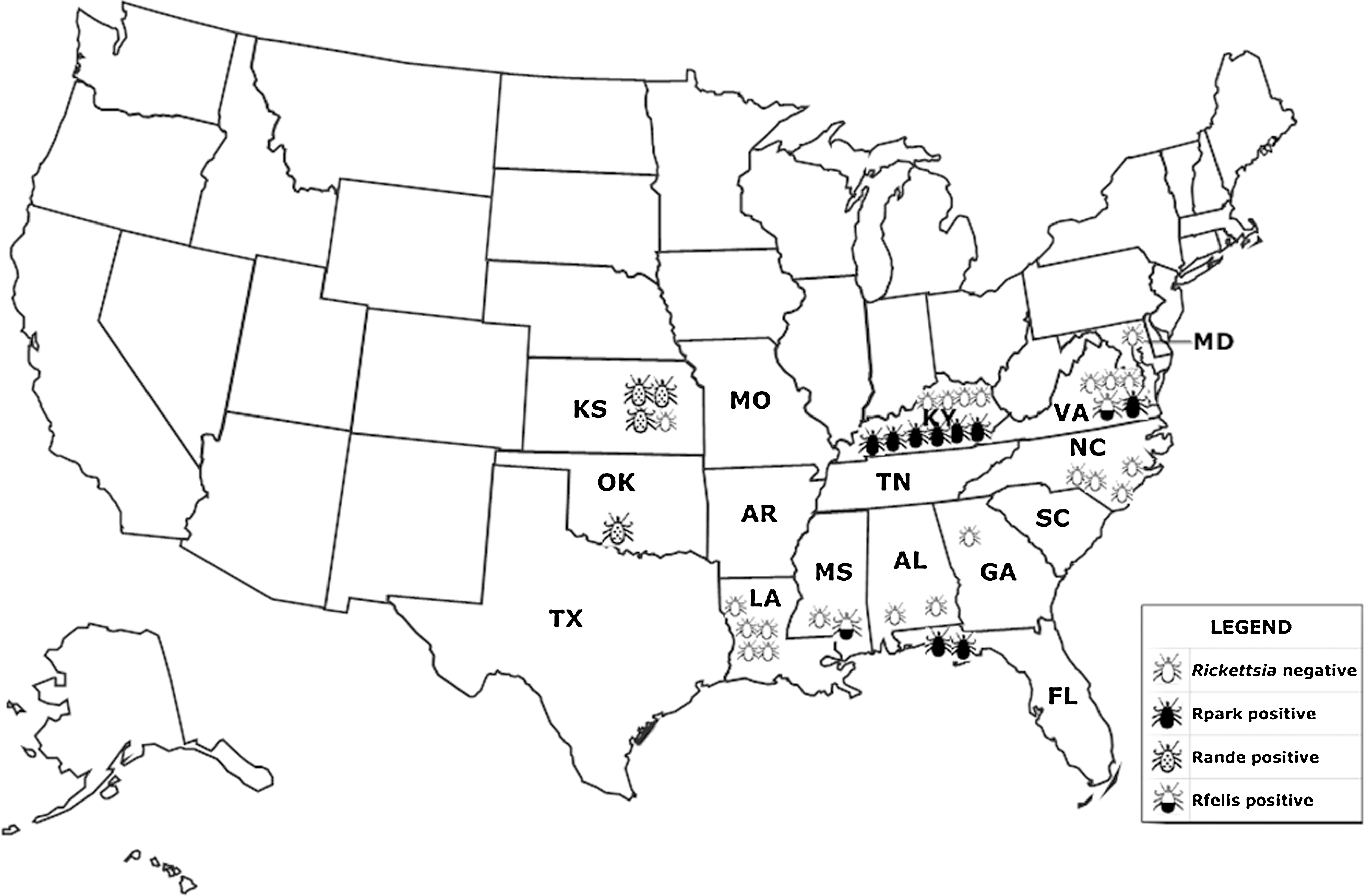
Map of Amblyomma maculatum endemic region with locations of Rickettsia parkeri and Candidatus Rickettsia andeanae infected Gulf Coast ticks.
Samples 1 and 2 were from individuals ticks found questing; samples 3–37 were collected from DoD personnel or their dependents reporting to a DoD health care clinic.
nd, not done; Ct: the number of cycles required to produce a fluorescence signal above the threshold level. The lower the Ct value, the greater the amount of target nucleic acid in the original sample. n, number of individual ticks included in the sample; ID, identification number/name for the samples; PCR/RFLP: polymerase chain reaction/restriction fragment length polymorphism analysis; USACHPPM; U.S. Army Center for Health Promotion and Preventive Medicine.
To assess the validity of using the Rande qPCR assay to detect Candidatus R. andeanae DNA, two nucleic acid preparations known to be positive for Candidatus R. andeanae DNA by MLST (Blair et al. 2004a, Jiang et al. 2005) (T124 and T163) were evaluated. In addition, two other nucleic acid preparations known to be SFG rickettsiae positive (Blair et al. 2004a) were also evaluated: one from an A. maculatum and the other from an Anocenter nitens (tick numbers T47 and T127, respectively). The two Candidatus R. andeanae positive samples were positive in the Rande qPCR assay, and one of the two SFG positive samples (T47) was also positive in the Rande assay. Two other tick samples from this study that were negative for evidence of infection with rickettsiae (Blair et al. 2004a) were accordingly negative by the Rande qPCR assay (data not shown).
To investigate the presence of Candidatus R. andeanae in the United States, the 37 A. maculatum tick preparations used to validate the Rpark assay were assessed by the Rande assay. Four of these samples representing individual ticks removed from humans in Kansas (n=3) and Oklahoma (n=1) (Fig. 1) were positive for both the Rick17b and the Rande qPCR assays (Table 3). Sequences of the sca4 from the four Rande positive nucleic acid preparations confirmed the Rande results and showed that these sequences (845 bp) were identical to those described for Candidatus R. andeanae molecular isolates T163 (GenBank accession No. GU395298) and T124 (Jiang et al. 2005). Thus, the Rande qPCR assay is capable of detecting Candidatus R. andeanae in ticks from both North and South America. Nine nucleic acid preparations that were positive for R. parkeri were negative for the Rande qPCR assay (Table 3), thus indicating that no R. parkeri and Candidatus R. andeanae coinfections were detected in this study.
Two tick DNA preparations were positive by the Rick17b qPCR assay but negative by the Rpark and Rande qPCR assays (samples 1 and 35). Sequence data from sca4 showed that sample 1, a single questing A. maculatum from Harborton, Accomack Co., VA, and sample 35, a single A. maculatum removed from a human from Hattiesburg, Forrest Co., MS, contained Rickettsia felis (100% identity of 877 bp of sca4 to R. felis URRWXCal2 [GenBank accession # CP000053]). These two samples were subsequently tested with the species-specific Rfelis qPCR assay, which targets a fragment of ompB (Henry et al. 2007) and confirmed the identification of R. felis DNA within the tick nucleic acid preparations (data not shown).
Discussion
This article describes the successful development, optimization, and sensitivity and specificity testing of three new quantitative real-time PCR assays, Rpark, Rande, and Rick17b. Analytical specificities of Rpark, Rande, and Rick17b qPCR assays were ascertained to be 100% by testing 32 nucleic acid preparations. In addition, the assays were found to have a consistent LOD of three copies per reaction. Thus, these assays should be invaluable for the use in detecting the human pathogen-R. parkeri, the newly identified rickettsia of unknown pathogenicity-Candidatus R. andeanae, and rickettsiae in general-Rickettsia spp. in clinical and environmental samples.
R. parkeri has been detected in A. maculatum ticks from Alabama, Arkansas, Florida, Georgia, Kentucky, Mississippi, Oklahoma, South Carolina, Texas, and Virginia in the United States (Parker et al. 1939, Sumner et al. 2007, Cohen et al. 2009, Paddock et al. 2010, Trout et al. 2010, Edwards et al. 2011, Wright et al. 2011) and from Piura in Peru (Florin et al. 2011); from Amblyomma triste ticks collected in Uruguay, Brazil, and Argentina (Venzal et al. 2004, Sangioni et al. 2005, Pacheco et al. 2006, Silveira et al. 2007, Nava et al. 2008); and in Amblyomma nodosum from Brazil (Ogrzewalska et al. 2009). In addition, a recent report described the detection of R. parkeri from an Amblyomma americanum tick in Tennessee and one in Georgia, raising concerns that this highly prevalent tick species may also be a vector for the pathogen, R. parkeri (Cohen et al. 2009). In the evaluation of Gulf Coast ticks from 11 southern states, we have detected for the first time evidence of R. parkeri in A. maculatum ticks parasitizing humans in Kentucky, 2003–2009, Virginia, 2008, and Florida, 2008. These sites are within the known range of R. parkeri infected A. maculatum (Sumner et al. 2007) and Tidewater spotted fever (Paddock et al. 2008).
The Rpark qPCR assay performed well not only with environmental samples but also with clinical samples, thus indicating its potential use in laboratory diagnosis of Tidewater spotted fever. This assay will be especially important in distinguishing Tidewater spotted fever from other American rickettsioses such as RMSF, flea-borne spotted fever, rickettsialpox, and epidemic and murine typhus (Azad et al. 1997, Paddock et al. 2006, Chapman et al. 2009, Labruna 2009, Parola et al. 2009, Zavala-Castro et al. 2009). Rpark qPCR assay could also be valuable in evaluation of future treatment and vaccine candidates against Tidewater spotted fever.
The Rande qPCR assay likewise has shown its utility in epidemiological investigations of rickettsial agents. The assay has successfully detected the presence of Candidatus R. andeanae in nucleic acid preparations of A. maculatum ticks both from Peru and the United States. This is the first time that the presence of Candidatus R. andeanae infected Gulf Coast ticks have been obtained from humans and the first description of the infected ticks from Kansas and Oklahoma. The only other reference to the presence of R. andeanae infected ticks reported in the United States was by Paddock et al. (2010) with two and three questing infected adult Gulf Coast ticks reported from Florida and Mississippi, respectively. Currently, it is unknown as to what effect Candidatus R. andeanae infected ticks might have on human health, but as has been the case with other tick-borne rickettsiae such as R. parkeri in the United States, R. slovaca and R. massiliae in Europe and R. heilongjiangensis in Asia, identification of new human rickettsioses followed decades later the discovery of the causative rickettsial agents in ticks (Paddock et al. 2004, Parola et al. 2005).
R. felis is the causative agent of flea-borne spotted fever, an emerging zoonotic disease. After its first identification in the cat flea in 1990 (Adams et al. 1990), this pathogen has been detected not only in its primary vector, the cat flea, but also in numerous arthropod species, including other species of fleas, ticks, and mites, with worldwide distribution in 28 countries over five continents (Reif et al. 2009). In addition, R. felis was detected in our lab from a Dermacentor variabilis removed from a human and submitted to the DoD Human Tick Test Kit Program from PA in 2003 (unpublished data). Herein, we report the first detection of R. felis in two A. maculatum ticks, one was an unfed male tick crawling on a dog in VA and the other was a female tick removed from a human in MS.
In conclusion, we describe the development of two species-specific real-time PCR assays, Rpark and Rande, that were successfully used to detect R. parkeri and Candidatus R. andeanae in A. maculatum ticks parasitizing humans in previously unreported locations in the United States.
Footnotes
Acknowledgments
The work reported herein was supported by the AFHSC-GEIS program, and its work unit number was 0000188M.0931.001.A0074. The opinions and assertions contained in this article are the private ones of the authors and are not to be construed as official or reflecting the views of the Navy Department, Army Department, or the Department of Defense at large. The authors, as employees of the U.S. Government, conducted the work as part of their official duties. Title 17 U.S.C. § 105 provides that “Copyright protection under this title is not available for any work of the U.S. Government.” Title 17 U.S.C § 101 defines a U.S. Government work as a work prepared by an employee of the U.S. Government as part of the person's official duties. The authors thank Mary Vince at USACHPPM for her technical help.
Disclosure Statement
No competing financial interests exist.