
Abstract
Crimean-Congo hemorrhagic fever (CCHF) is a virulent tick-borne disease with a case fatality rate ranging from 10–50% for tick-borne transmission, and up to 80% for nosocomial transmission. Human cases have been reported in over 30 countries across Europe, Asia, and Africa. It appears to be spreading to new areas with several countries reporting their first human cases of CCHF disease within the past 10 years. We report a novel real-time RT-PCR assay designed to amplify a conserved region of the CCHF virus S segment. It is capable of detecting strains from all 7 groups of CCHF, including the AP92 strain that until recently represented a lineage of strains that were not associated with human disease. The limit of detection of the assay is 5 copies of target RNA, and the assay shows no cross-reactivity with other viruses from within the same genus, or with viruses causing similar human disease.
Introduction
The CCHF virus (CCHFv) genome is comprised of single-stranded negative-sense RNA divided into 3 distinct segments designated small (S), medium (M), and large (L). The L segment encodes the RNA-dependent RNA polymerase, the M segment encodes the precursor of the two envelope glycoproteins Gn and Gc, and the S segment encodes the nucleocapsid protein. The virus is maintained in nature through a transmission cycle involving both ticks and vertebrate hosts, and there is a strong correlation between distribution of the predominant tick vector (Hyalomma marginatum marginatum) and the incidence of human disease (Hoogstraal 1979).
Humans appear to be the only known host, apart from newborn mice, in which disease is manifested (Whitehouse 2004). In contrast to the asymptomatic infection of other vertebrate hosts, human infection is frequently typified by severe hemorrhagic disease that often results in a fatal outcome. A common route of human infection is via the bite of an infected tick, and the probability of developing disease via this route has been estimated to be approximately 20% (Goldfarb et al. 1980). In addition to infection via tick bite, transmission via contact with infected tissue and fluids is also possible, and nosocomial outbreaks have been well documented in several countries across the world (Burney et al. 1980; van Eeden et al. 1985; Harxhi et al. 2005; Fisher-Hoch 2005; Gurbuz et al. 2009; Mardani et al. 2009; Aradaib et al. 2010). The clinical course of the disease is widely described, and is typified by four distinct phases: incubation, prehemorrhage, hemorrhagic, and in patients who recover, convalescence. The duration of each phase is highly variable. An asymptomatic incubation period lasting 1–3 days leads to a prehemorrhage phase characterized by sudden onset of fever (39–41°C), headache, and myalgia. Hemorrhagic manifestations such as petechiae and ecchymosis mark the start of the hemorrhagic phase, and bleeding can occur from numerous sites including skin, mucous membranes, gingiva, nose, respiratory tract, gastrointestinal tract, uterus, urinary tract, and cerebrum. For non-fatal disease, convalescence begins 2–3 weeks after the onset of symptoms, and may include long-term sequelae such as nausea, impaired vision, and minor neurological impairment, although these conditions are rarely permanent (Hoogstraal 1979; Whitehouse 2004; Ergonul 2006; Mardani and Keshtkar-Jahromi 2007).
CCHF is the most widely distributed tick-borne disease, and to date has been reported in more than 30 countries throughout Africa, Asia, and Europe (Mardani and Keshtkar-Jahromi, 2007). Several recent reports have highlighted the importance of surveillance to monitor the potential spread of CCHF into new areas (Senior 2008; Havelaar et al. 2010; Maltezou and Papa, 2010). Significantly, within the past 10 years confirmed clinical cases of CCHF have been recorded for the first time in Kenya, Turkey, Greece, Sudan, Georgia, and India (Dunster et al. 2002; Karti et al. 2004; Papa et al. 2009; Aradaib et al. 2010; Mishra et al. 2011; Zakhashvili et al. 2010).
The relentless spread and/or increased detection of clinical cases highlight the need for rapid and reliable diagnostic methods to effectively detect the presence of CCHFv. To date, several real-time RT-PCR assays have been described for the detection of CCHFv (Yapar et al. 2005; Duh et al. 2006; Garrison et al. 2007; Wolfel et al. 2007); however, these collectively suffer from drawbacks, including multi-probe detection format (increased expense), limited geographical targeting (decreased specificity), or lack of validation. The objective of this study was to design a novel, single-probe, real-time RT-PCR assay capable of detecting all known strains of CCHFv, including genetically diverse strains that have only recently been associated with human disease (Midilli et al. 2009; Ozkaya et al. 2010).
Materials and Methods
Primer and probe design
Comparison of published S segment sequences from isolates collected from across Europe, Asia, and Africa show a high degree of variability (Drosten et al. 2002; Hewson et al. 2004; Chamberlain et al. 2005; Han and Rayner, 2011). Thus, it is of great importance that any diagnostic assay has the capability to detect the complete global spectrum of isolates. The conservation of complementary nucleotide sequences at the 5′ and 3′ termini is a feature seen in all genera within the family Bunyaviridae, and it is through this that the base-pairing between these two regions forms a “pan-handle” structure which is involved in recognition by the viral polymerase (Flick et al. 2002). A forward primer was designed to this highly conserved 5′ untranslated region of the S segment, and the reverse primer was selected by comparing all available S segment sequences on GenBank and selecting the most homogeneous region based on nucleotide and protein sequence using these data. The reverse primer was also designed to bind to a conserved tryptophan residue at the 3′ terminus of the priming sequence, ensuring that the crucial 3′ end of the primer binds to sequence with only one potential codon.
A single probe capable of binding to all published strains with only a single degeneracy was designed within this amplicon. This probe was designed with a 5′6-carboxifluorescein (FAM) fluorophore and a 3′ Black Hole Quencher 1 (BHQ1) quencher to maximize potential sensitivity by reducing background fluorescence (Yang et al. 2009).
As shown in Table 1, the sequence of the forward primer (designated CCHF S1) is TCT CAA AGA AAC ACG TGC C, the sequence of the reverse primer (designated CCHF S122) is CCT TTT TGA ACT CTT CAA ACC, and the sequence of the probe (designated CCHF probe) is FAM-ACT CAA GGK AAC ACT GTG GGC GTA AG-BHQ1. All sequences are shown in the 5′ to 3′ orientation.
Representative sequence: the underlined region is sequence-altered from wild-type.
To use the contamination control probe in the CCHF real-time RT-PCR assay, add 0.2 μL of 25 μM stock to the master mix documented in the materials and methods section, and reduce the volume of water in order to maintain a 20-μL reaction volume. To interpret the results, run the initial PCR on the 530 nm (FAM) channel as standard, then switch to the 560 nm (JOE) channel after completion to assess whether positives also fluoresce at this wavelength; fluorescence at 560 nm indicates that contamination with the synthetic control has occurred. NB color compensation must be carried out to reduce bleed-through from the FAM reporter.
Virus propagation and RNA isolation
Viral RNA from 16 unique strains of CCHF virus were used for the evaluation process (Table 2). Viruses were cultured in vitro using Vero E6 cells and L15 medium supplemented with 2% serum. Virus propagation was performed under high containment (Containment Level 4) conditions. Infectious virus was inactivated using AVL buffer (Qiagen Inc., Valencia, CA), and viral RNA was purified using the QIAamp viral RNA mini kit (Qiagen), in accordance with the manufacturer's instructions. RNA from strains 87/07, Congo 1976, 39554, BT958, and Mg954 was obtained through an external quality assurance program, which was part of an independent project.
Based on Hewson et al. 2004.
As described in Garrison et al. 2007.
As described in Wölfel et al. 2007.
RNA transcripts.
Figures in brackets refer to Crossing Point values as indicates by LightCycler software version 4.0 (for LightCycler 2.0), or software version 1.5 (for LightCycler 480).
EQA, limited RNA stocks provided as part of FP7 External Quality Assurance programme; In Progress, submission to Genbank in progress.
These 16 strains covered 5 of the 7 genomic lineages described (Hewson et al. 2004). Synthetic RNA for representative members of the two remaining lineages was produced according to the GenBank sequences (AP92 strain from lineage Europe 2, accession number DQ211638, and DAK 8194 strain from lineage Africa 1, accession number U88411). S segment synthesis was carried out by a commercial company (GeneArt®, Invitrogen, Carlsbad, CA), and RNA transcripts of the entire S segment were carried out using the MEGAscript kit (Ambion, Austin, TX) in accordance with the manufacturer's instructions.
Real-time RT-PCR
The real-time RT-PCR assay was developed and validated using the LightCycler 2.0 and LightCycler 480 platforms (both Roche Diagnostics Corp., Indianapolis, IN), and SuperScript III (SSIII) Platinum One-step qRT-PCR kit (Invitrogen). The final master mix (15 μL) comprised 10 μL of 2× Reaction Mix, 1.7 μL of PCR-grade water, 1 μL of each primer (at 18 μM working concentration), 0.5 μL of probe (at 25 μM working concentration), and 0.8 μL of SSIII enzyme mix. Then 5 μL of template RNA was added to the master mix in order to give a final reaction volume of 20 μL. The cycling conditions used were 50°C for 10 min, 95°C for 2 min, followed by 45 cycles of 95°C for 10 sec and 60°C for 40 sec (with quantification analysis of fluorescence performed at the end of each 60°C step), and a final cooling step of 40°C for 30 sec.
Synthetic positive control material
A synthetic RNA-transcript positive control can be implemented in this assay to ensure the validity of diagnostic positives, for example when transitioning to new laboratories and/or operators. This control is based upon the 122-base-pair amplicon produced by known sequences of CCHFv, with a section of the non-primer/probe binding region altered to produce an atypical sequence. A second probe was designed against this altered region, with a 6-carboxy-4′,5′-dichloro-2′,7′-dimethoxyfluorescein (JOE) fluorophore utilized to allow detection of fluorescence outside of the fluorescence range used for the primary (FAM) probe. This secondary probe will only detect this specific modified positive control, and utilization of this control will allow for in-run monitoring of potential laboratory contamination. A dilution series of this synthetic RNA-transcript control can also be used to produce standard curves and estimation of copy number.
Table 1 documents the sequence of the synthetic control in comparison to a wild-type strain,a and the amendments necessary to incorporate this control in routine testing.b Synthetic control DNA was produced commercially (GeneArt), and transcript RNA of this sequence was produced using the MEGAshortscript T7 kit (Ambion) in accordance with the manufacturer's instructions.
Results
Specificity and cross-reactivity testing
All 18 strains of CCHFv listed in Table 2 were detected by using this novel real-time RT-PCR. These strains represent all 7 lineages described previously (Hewson et al. 2004), including the Europe 2 lineage that contains the AP92-like strains.
RNA purified from a broad spectrum of viral pathogens was run using this assay. No false-positives were detected. This panel included several viruses from the Bunyaviridae family to demonstrate specificity to CCHFv, as well as other viruses that have the ability to cause hemorrhagic fevers (Table 3).
Viral RNA obtained from the HPA Special Pathogens Reference Unit; 5 μL of sample was used at standard dilution as positive control material in diagnostic testing.
This methodology is transferrable between several real-time platforms, and has been validated on the LightCycler 480 and LightCycler 2.0 (both Roche Diagnostics), ABi 7500 Fast and 7900 Fast (both Applied BioSystems, Inc., Carlsbad, CA), SmartCycler (Cepheid, Sunnyvale, CA), and MiniOpticon (BioRad, Inc., Hercules, CA) platforms utilizing the same reaction and cycling conditions (Table 4).
Assay performance on six different real-time platforms.
Copy number based on RNA transcripts of the entire S segment of AP92. All values represent Crossing Point (CP)/Crossing Threshold (Ct)/Quantification Cycle (Cq) data as produced by platform software (data averaged across replicates, and then rounded to the nearest whole number).
No platform tested was able to detect 100% of replicates for 5 copies per reaction. All replicates of 50 copies per reaction (and higher dilutions) were detected.
Comparison with previously published assays
Two widely used published CCHF real-time RT-PCR assays (Garrison et al. 2007; Wolfel et al. 2007) were tested against the panel of 18 CCHFv strains in parallel with the assay described in this report. Results are shown in Table 2. While all three assays detected the vast majority of strains, only the assay reported here was able to detect the AP92 and DAK 8194 CCHFv strains. Alignments of these two strains against primer and probe sequences (Fig. 1) show a high degree of mismatch, in particular at the crucial 3′ end of the primers, for both the previously published assays, possibly explaining the lack of amplification. The majority of mismatches shown in Figure 1 are non-critical due to the minor effects of purine-pyrimidine base mismatching (Stadhouders et al. 2010); however, both the contemporary assays contain more critical purine-purine and/or pyrimidine-pyrimidine mismatches when compared against certain strains. While the presence of a single mismatch will not necessarily result in a lack of amplification, it is likely that the cumulative effect of minor mismatches, in combination with these more critical mismatches throughout both the primer and probe binding regions, could result in the failure to amplify the AP92 and DAK 8194 strains, especially if there are mismatches in the last few bases of the 3′ end of either primer.

(
In general the Crossing Point (CP) values generated by the 3 assays were relatively consistent, with values rarely varying by more than 1 or 2 cycles, indicating similar sensitivities between the assays; however, there were some strains that showed a marked increase in detection by using the assay described in this article, notably strains 39554, BT958, and D8692, all of which were detected at least 4 cycles earlier in comparison to the two published assays.
Limit of detection
Production of a synthetic S segment provided quantifiable genetic material to estimate the limit of detection. Evaluation of transcript RNA concentration was achieved using nanodrop technology (Thermo Scientific, Fremont, CA), and a 10-fold dilution of this material was performed. Seven dilutions of the AP92 transcript RNA were tested against the new assay, ranging from 5×105 copies of S segment RNA per reaction, down to 0.5 copies of S segment RNA per reaction (Fig. 2). Using 10-fold dilutions of the starting material, the limit of detection of CCHFv using this assay was estimated to be 5 S segment copies of RNA per PCR reaction (3 of 4 replicates detected). In-run analysis (LightCycler 480 software version 1.5) indicated the reaction efficiency to be 2.09, with a slope of −3.12, a Y intercept of 39.17, and the R2 value of the reaction was determined to be greater than 0.99.
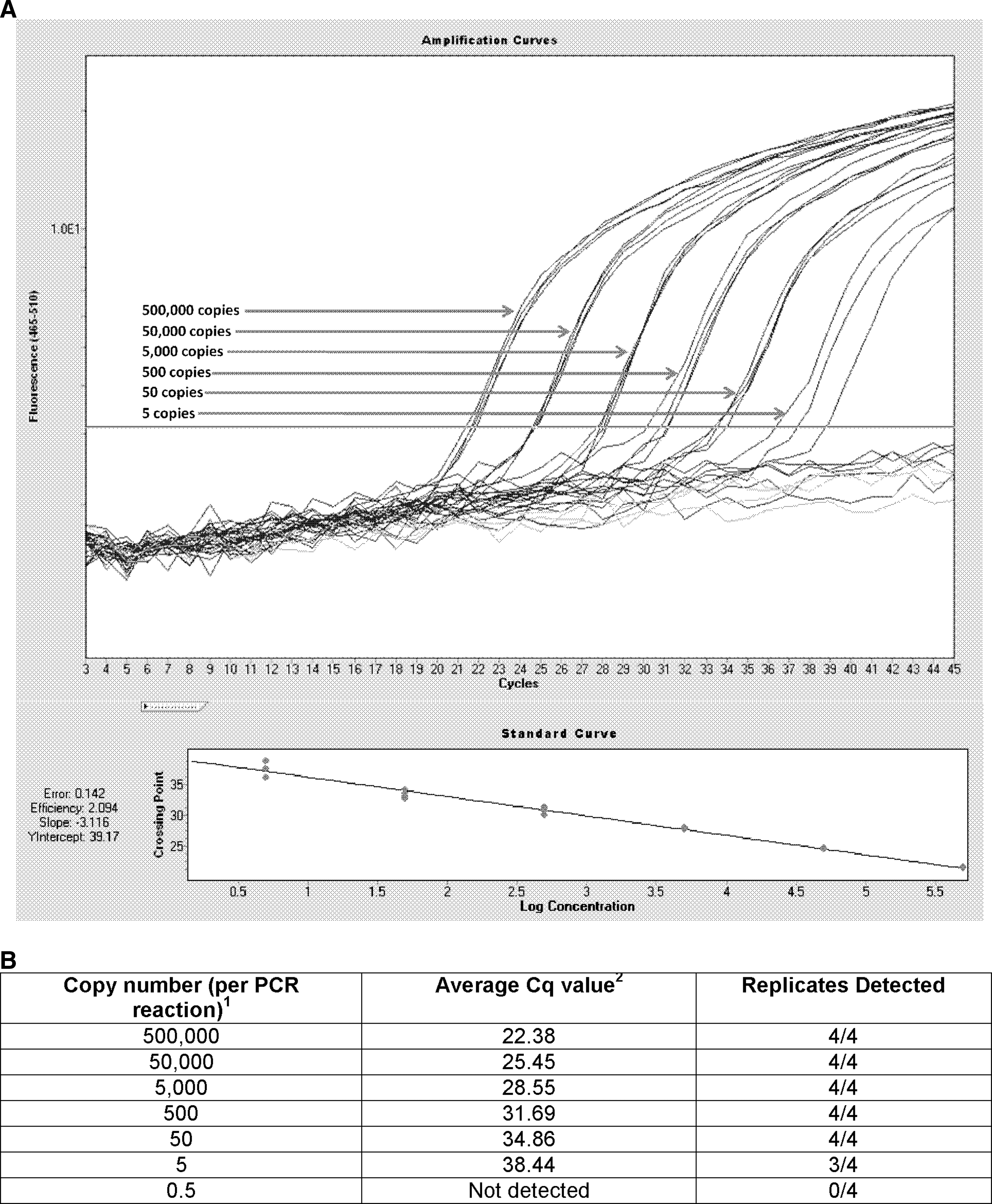
(
Clinical application
After full validation, this assay has been introduced and used in a clinical setting within the Special Pathogens Reference Unit (Health Protection Agency–U.K.). It has been used to diagnose clinical cases during CCHF outbreaks in Pakistan, Kosovo, and Tajikistan (manuscripts in preparation).
Discussion
CCHF is an important zoonosis and human pathogen. Over 30 countries worldwide have reported human cases of the disease, with several countries having recognized CCHF as a novel endemic disease in recent years. The apparent increase in the global distribution of this pathogen, coupled with the significant increase in case fatality rates associated with nosocomial transmission of the virus, highlight the importance of a rapid, robust, and reliable diagnostic procedure to confirm the presence of the virus. Assays that lead to early diagnosis of CCHF will result in better patient management and isolation and infection control.
The real-time RT-PCR assay described in this article was able to detect all strains of CCHFv available for testing. The assay performed favorably when compared to two previously validated CCHFv real-time RT-PCR assays (Garrison et al. 2007; Wolfel et al. 2007), and in general offers greater specificity and sensitivity than either of these previous reports. The limit of detection was determined to be 5 copies of S segment RNA with a 75% detection rate, and all replicates of higher dilutions were successfully detected. The cost of running this assay is also considerably less than the two previously reported assays; the single FAM-BHQ1 probe results in lower reagent costs in comparison to a multiple-probe or Minor Groove Binder format. The PCR kit list price for this assay is currently more than 25% cheaper per 100 reactions than the other published assays.
AP92 is unique among strains of CCHFv, both in terms of genetic make-up and clinical outcome. AP92 is considered to cause a much milder form of human disease, and may represent predominantly asymptomatic infections (Antoniadis and Casals, 1982; Ozkaya et al. 2010). The genetic diversity from other more pathogenic strains has been problematic in terms of designing pan-CCHFv assays, and their necessity could be questioned due to a lack of clinical cases. However, recent reports of AP92-like strains of CCHF causing human illness (Midilli et al. 2009; Ozkaya et al. 2010) highlight the importance of having a robust assay that can detect the strains currently considered to be apathogenic.
By designing the PCR primers to a conserved region of the CCHFv S segment it was possible to create a simple, single-probe assay capable of amplifying target RNA from all strains of CCHFv available for analysis. Due to the highly conserved nature of the extreme terminal regions of the S segment it is expected that an assay designed to target this region will be more likely to detect all strains of the virus, due to a constrained sequence context and reduced pressure for genetic variation.
To our knowledge, this is the first single-probe real-time PCR assay capable of detecting strains from all 7 groups of CCHFv, and offers a simple, cost-effective diagnostic assay for the detection of CCHFv.
Utilization of a plasmid control to estimate copy number, and/or the modified positive control template to identify potential laboratory contamination, can further increase the reliability of this assay, although neither is essential for diagnostic purposes.
Footnotes
Acknowledgments
The authors would like to acknowledge the assistance of Prof. Farida Tishkova (Tajik Science and Research Institute of Preventive Medicine, Dushanbe, Tajikstan), and Prof. Salih Ahmeti (Clinic of Infectious Diseases, Prishtina, Republic of Kosovo/Serbia) in providing strains of CCHFv used in this evaluation.
Author Disclosure Statement
This report contains work commissioned by the National Institute for Health Research. The views expressed are those of the authors and not necessarily those of the NHS, the National Institute for Health Research or the Department of Health.