
Abstract
Recent reports of novel hantaviruses in shrews and moles and the detection of rodent-borne hantaviruses in different rodent species raise important questions about their host range and specificity, evolution, and host adaptation. Tula virus (TULV), a European hantavirus, is believed to be slightly or non-pathogenic in humans and was initially detected in the common vole Microtus arvalis, the East European vole M. levis (formerly rossiaemeridionalis), and subsequently in other Microtus species. Here we report the first multiple RT-PCR detection and sequence analyses of TULV in the Eurasian water vole Arvicola amphibius from different regions in Germany and Switzerland. Additional novel TULV S-, M-, and L-segment sequences were obtained from M. arvalis and M. agrestis trapped in Germany at sites close to trapping sites of TULV-RT-PCR-positive water voles. Serological investigations using a recombinant TULV nucleocapsid protein revealed the presence of TULV-reactive antibodies in RT-PCR-positive and a few RT-PCR-negative water voles. Phylogenetic analyses revealed a geographical clustering of the novel S-, M-, and L-segment sequences from A. amphibius with those of M. arvalis- and M. agrestis-derived TULV lineages, and may suggest multiple TULV spillover or a potential host switch to A. amphibius. Future longitudinal studies of sympatric Microtus and Arvicola populations and experimental infection studies have to prove the potential of A. amphibius as an additional TULV reservoir host.
Introduction
On the Eurasian continent hantaviruses may cause hemorrhagic fever with renal syndrome (HFRS) of different severity and case fatality rates of up to 10% (Krüger et al. 2011). One representative is the bank vole Myodes glareolus-transmitted Puumala virus (PUUV) distributed in almost all parts of Europe and causing a mild to moderate form of HFRS, designated nephropathia epidemica, with a case fatality rate of <0.1% (Brummer-Korvenkontio et al. 1999). Second, Dobrava-Belgrade virus (DOBV) genetic lineages DOBV-Af, DOBV-Ap, DOBV-Aa, and Saaremaa associated with the yellow-necked mouse Apodemus flavicollis, Caucasian wood mouse A. ponticus, or striped field mouse A. agrarius cause more severe, moderate, or mild/moderate cases of HFRS in certain parts of Europe, respectively (Avsic-Zupanc et al. 1999; Sibold et al. 2001; Vapalahti et al. 2003; Golovljova et al. 2007; Klempa et al. 2008; Krüger et al. 2011).
The knowledge of the human pathogenicity of the third hantavirus in Europe, the Tula virus (TULV), is sparse; there are only few reports of human TULV infections (Vapalahti et al. 1996; Schultze et al. 2002; Clement et al. 2003; Klempa et al. 2003; Mertens et al. 2011). This hantavirus has initially been found in the common vole Microtus arvalis, the East European vole M. levis (formerly rossiaemeridionalis), and subsequently in M. agrestis, M. subterraneus, and M. gregalis, and is broadly distributed in Central Europe (Plyusnin et al. 1994; Sibold et al. 1995; Song et al., 2002; Scharninghausen et al., 2002; Schmidt-Chanasit et al. 2010).
In general, the genome of hantaviruses is represented by three single-stranded RNA genome segments of negative polarity. The structural proteins of the hantaviruses, the nucleocapsid (N) protein, and the glycoproteins Gn (G1) and Gc (G2), are encoded by the small (S) segment of 1.6–2.0 kilobases (kb), and the medium (M) segment of 3.5–3.6 kb, respectively. The RNA-dependent RNA polymerase is encoded by the large (L) segment of approximately 6.5 kb.
The close association of a single hantavirus species with a single reservoir or closely related species of the same genus has been explained by a co-evolution hypothesis (Plyusnin and Morzunov 2001). However, the increasing number of hantavirus species, hantavirus studies on sympatrically-occurring rodent reservoir species (Schmidt-Chanasit et al. 2010), and the discovery of insectivore-borne hantaviruses in particular raises major questions on the evolution and host adaptation of hantavirus species (Henttonen et al. 2008). Alternatively to the virus-host co-evolution, recent studies have postulated a scenario of host switching and local host-specific adaptation for hantavirus/host evolution (Ramsden et al. 2008). Further, a host switch event in the distant past has been postulated for the ancestor of the Arvicolinae-associated Khabarovsk virus (Vapalahti et al. 1999).
The Eurasian water vole (Arvicola amphibius, formerly A. terrestris), like Microtus, is a member of the subfamily Arvicolinae, and is widely distributed in Europe. Interactions or sympatric occurrences of water voles with other arvicolines (e.g., M. arvalis) have been described, and their fluctuations in the population density sometimes correlate with the population density in A. amphibius (Wieland 1973). So far investigations of hantavirus infections in A. amphibius are sparse. Hantavirus antigen was detected in lung samples of A. amphibius from Russia using immunoglobulin G (IgG) antibodies directed against PUUV, Hantaan virus, and Vladivostok virus (Butenko et al. 1997). PUUV-reactive antibodies were demonstrated in 5.5% of 164 montane water voles (Arvicola scherman) trapped in France (Charbonnel et al. 2008). In this study, we report the first molecular evidence of multiple TULV infections in A. amphibius from Germany and Switzerland.
Materials and Methods
Rodent trapping and necropsy
During 2001 to 2009 a total of 424 A. amphibius were trapped at 20 different sites in Germany and at one locality in Switzerland. In addition, six M. arvalis and one M. agrestis trapped at three sites in Germany were included in this study (Fig. 1). The animals were necropsied according to standard protocols (Ulrich et al. 2008).

Map of trapping sites with RT-PCR/serologically positive and negative Arvicola amphibius, Microtus arvalis, and M. agrestis (stars indicate trapping sites with seroreactive and RT-PCR positive/equivocal A. amphibius; circles with white centers indicate trapping sites with seroreactive but RT-PCR-negative A. amphibius; diamonds indicate trapping sites with RT-PCR equivocal and negative A. amphibius; circles with crosses in the center indicate trapping sites with RT-PCR negative A. amphibius; solid circles indicate trapping sites with Tula virus (TULV)-RT-PCR-positive M. arvalis and M. agrestis; “x” indicates the origin of published TULV sequences; Schmidt-Chanasit, et al. 2010). Trapping sites: Ben, Bendelin; Ebe, Eberswalde; Mrz, Marzehns; Bieb, Biebersdorf; Duer, Dürrweitzschen; Schar, Scharfenberg; Goett, Göettingen; Sen, Sennickerode; Wen, Wenigenlupnitz; Sieb, Siebleben; Eck, Eckhardtshausen; Graf, Grafenwöhr; Winn, Winnenden; Wies, Wiesensteig; Roem, Römerstein; Heu, Heuberg; Nof, Noflen.
RT-PCR and sequencing
Lung samples were investigated by two L-, one S-, and one M-segment-specific RT-PCR assays. The initial screening occurred through a newly established SYBR-Green-based real-time reverse transcription-PCR (RT-qPCR) assay using the QuantiTect SYBR Green RT-PCR Kit (Qiagen, Hilden, Germany) and novel degenerated primers (L2797F 5′ GAR GAR TAY ATH TCN TAT GGD GG-3′; L2951R 5′-HGG NGA CCA YTT NGT DGC AT-3′) targeting a conserved region in the L-segment (nt 2797-2819 and nt 2951-2970; positions according to NCBI reference sequence TULV, accession number: NC 005226) of different rodent- and insectivore-borne Old World hantaviruses. This assay was experimentally shown to detect DOBV-, PUUV-, and TULV-specific nucleic acid sequences in lungs from naturally-infected Apodemus, Myodes, and Microtus rodents from Germany (data not shown). Each water vole lung sample was determined as positive if the ct-value was <38 and by detection of a specific amplification product in a melting curve and in a 1.5% agarose gel. Samples with no ct-value and no specific amplification product were defined as negative, whereas those with a ct-value >38 and a typical melting curve were considered as equivocal. In addition, the samples from Baden-Wuerttemberg were tested in a nested RT-PCR assay using S-segment-specific primers (Sibold et al. 1999). All RT-qPCR positive and equivocal samples, as well as positive samples from the nested RT-PCR assay, were also tested in a One-Step RT-PCR assay using a Superscript III One Step RT-PCR Kit (Invitrogen, Darmstadt, Germany), and with primers targeting another region in the L-segment (Klempa et al. 2006). Thereafter, all L-segment-positive lung RNA samples were tested in S- and M-segment RT-PCR assays using primers specific for TULV and PUUV (Essbauer et al. 2006; Schmidt-Chanasit et al. 2010). The RT-PCR products were purified with a PCR Purification Kit (Qiagen), and sequenced using the BigDye terminator sequencing kit (Perkin-Elmer, Waltham, MA) on an ABI 310 Genetic Analyzer (Applied Biosystems, Foster City, CA).
Phylogenetic analysis
The phylogenetic analyses were performed using MrBayes 3.1.2 (Ronquist and Huelsenbeck 2003) with Bayesian Metropolis-Hastings Markov Chain Monte Carlo (MCMC) tree-sampling methods based on two MCMC runs consisting of four chains of 2,000,000 generations with a burn-in of 25%, and second by maximum-likelihood (ML) analysis calculated on a web server (
Serology
Serological screening of phosphate-buffered saline (PBS) diluted chest cavity fluid (CCF) was done by an in-house IgG ELISA using a yeast-expressed TULV nucleocapsid protein (Mertens et al. 2011), and by following a previously published protocol (Essbauer et al. 2006). The CCFs were diluted 1:10 in 0.5% bovine serum albumin/0.05% Tween-20. Horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (Bio-Rad, Hercules, CA) was used as secondary antibody. Detection of specific antibodies was accomplished by addition of 3,3′,5,5’-Tetramethylbenzidine (TMB) substrate (Bio-Rad). The reaction was stopped by the addition of 50 μL of 1 M H2SO4 (10 min).
Cytochrome b PCR
For all TULV-RT-PCR-positive A. amphibius from the seven trapping sites and one additional non-infected individual from each trapping site the morphological species determination was confirmed by a mitochondrial cytochrome b (cyt b) gene-specific PCR (Schlegel et al. 2011), and a subsequent BLAST search-mediated comparison of the novel cyt b sequences with sequences available in GenBank (
Results
An initial screening of all 424 Arvicola lung samples with the new SYBR-Green RT-qPCR assay identified 4 positive and 32 equivocal samples from eight different trapping sites in Germany and Switzerland (Table 1 and Fig. 1). The subsequent analysis of all 36 RT-qPCR-positive and equivocal samples and 2 RT-qPCR-negative, but previously positive screened samples (GER127 and GER129) in a One-Step RT-PCR assay with primers targeting another region in the L-segment (Klempa et al. 2006) revealed a total of 8 samples showing the expected amplification product in the agarose gel, but only for 7 samples could a sequence be obtained (Table 1). These sequences represent a 336-bp-long part of the L-segment (nt 2962–3297 encoding aa 988–1099 of the RNA polymerase).
Samples with the same ELISA reactivity and RT-PCR results.
Optical density values:+++, ≥2.8; +, 0.5–0.3; (+), 0.2–0.13; neg, <lower cut-off (on average 0.041).
Agarose gel analysis showed a weak amplification product of the expected size; sequencing approaches remained unsuccessful.
F, female; M, male; ND, not done; Neg, negative; TULV, Tula virus; Duer, Dürrweitzschen; Wen, Wenigenlupnitz; Roem, Römerstein; Winn, Winnenden; Wies, Wiesensteig; Schar, Scharfenberg; Nof, Noflen; Eck, Eckhardtshausen.
An initial BLAST search demonstrated the strongest similarity of these Arvicola-derived L-segment sequences to corresponding TULV sequences from M. arvalis and M. agrestis (data not shown). Therefore, we determined additional novel TULV partial L-segment as well as partial S- and M-segment sequences from M. arvalis and M. agrestis trapped in Baden-Wuerttemberg (Heu), Brandenburg (Bieb), and Thuringia (Sieb) near sites where positive A. amphibius have been previously identified (Fig. 1). A pair-wise comparison of the Arvicola-derived TULV L-segment and deduced RNA polymerase sequences revealed nucleotide and amino acid sequence differences of 14.6–21% and 0–5.5%, respectively. All 8 L-segment positive lung RNA samples were then tested in S- and M-segment RT-PCR assays using TULV- and PUUV-specific primers. The obtained RT-PCR products were sequenced and found to represent a 378-bp-long part of the M-segment (nt 2365–2742 encoding aa 789–914 of the glycoprotein precursor), and a 549-bp-long part of the S-segment (nt 379–927 encoding aa 127–305 of the N protein).
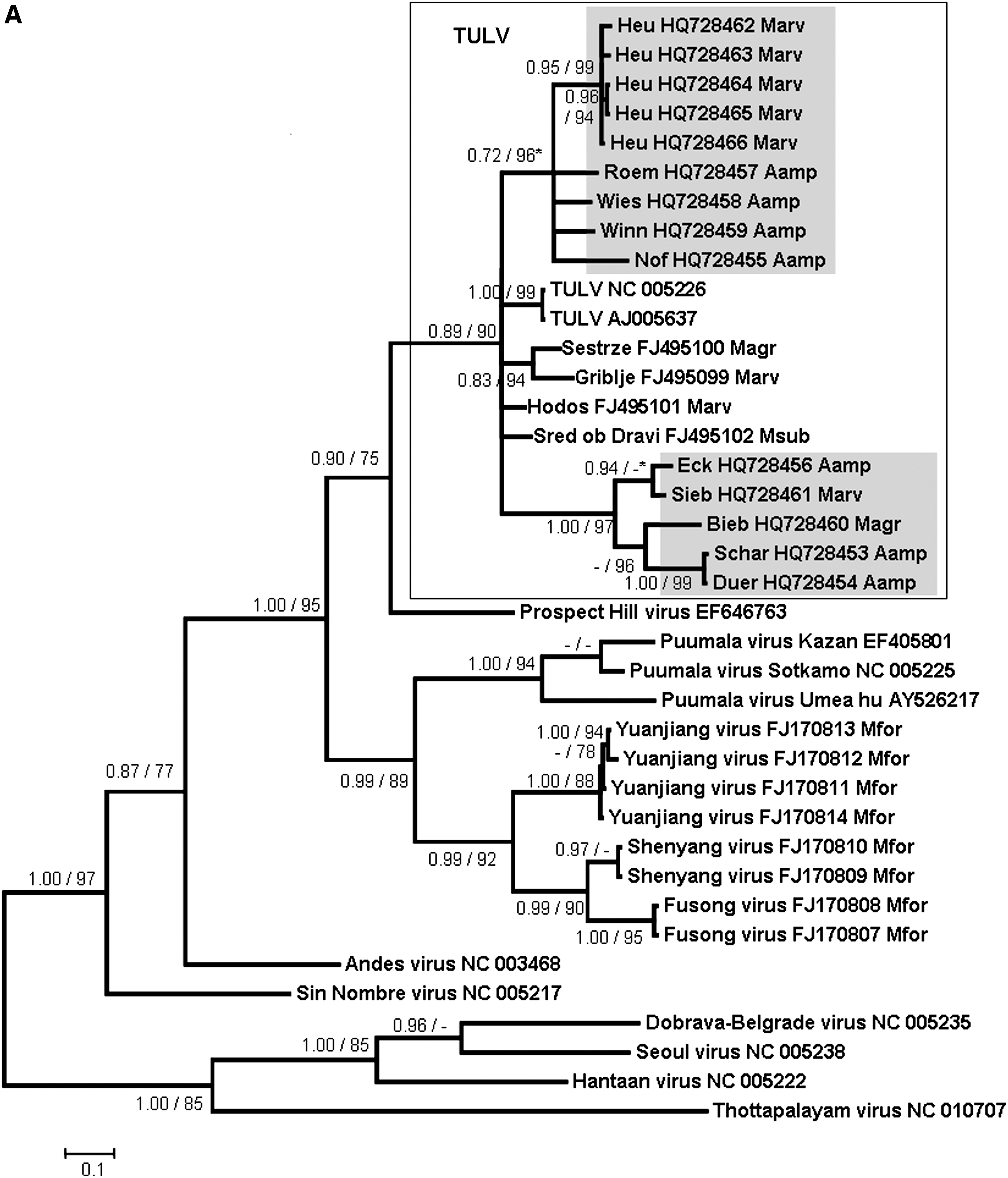
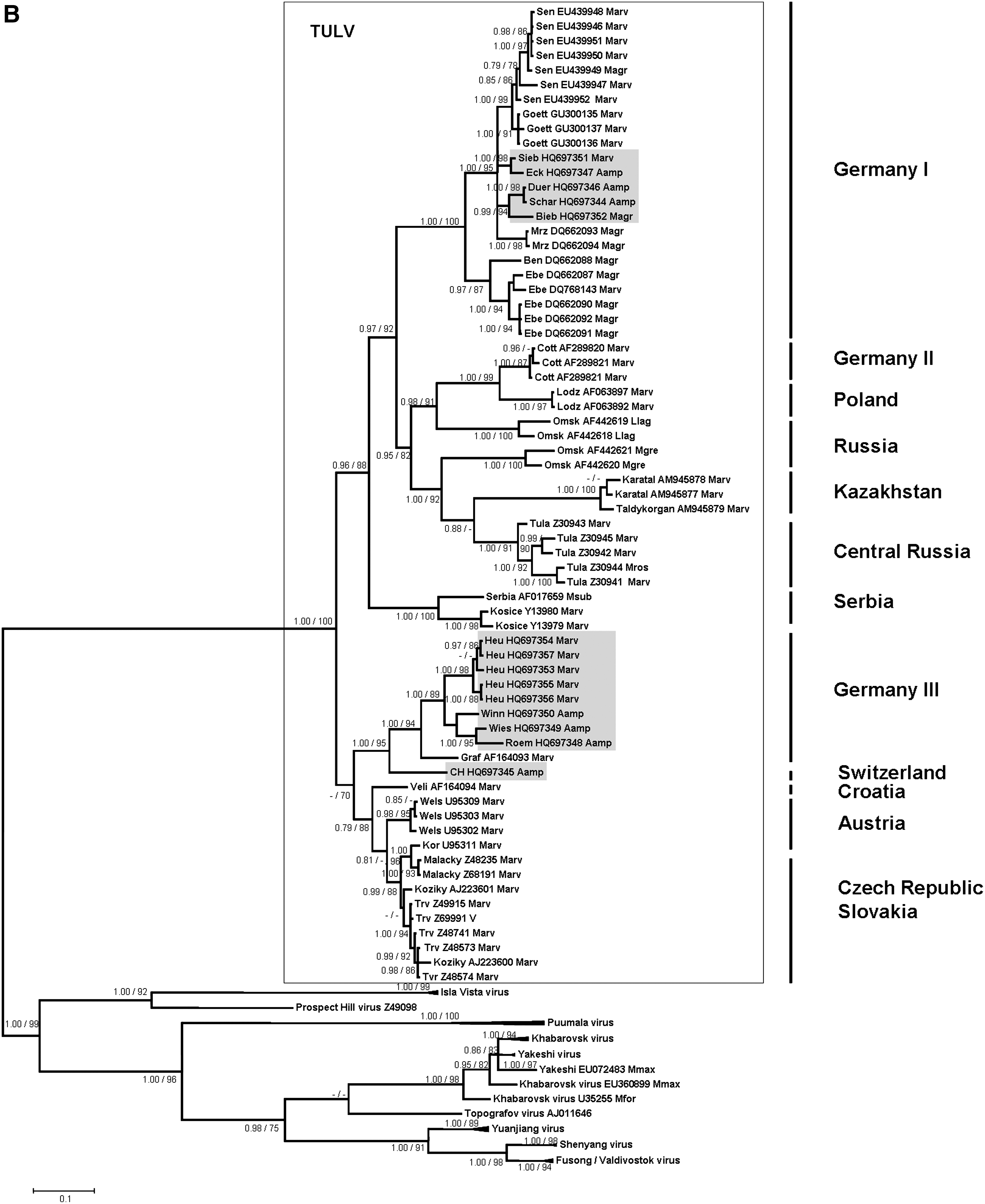
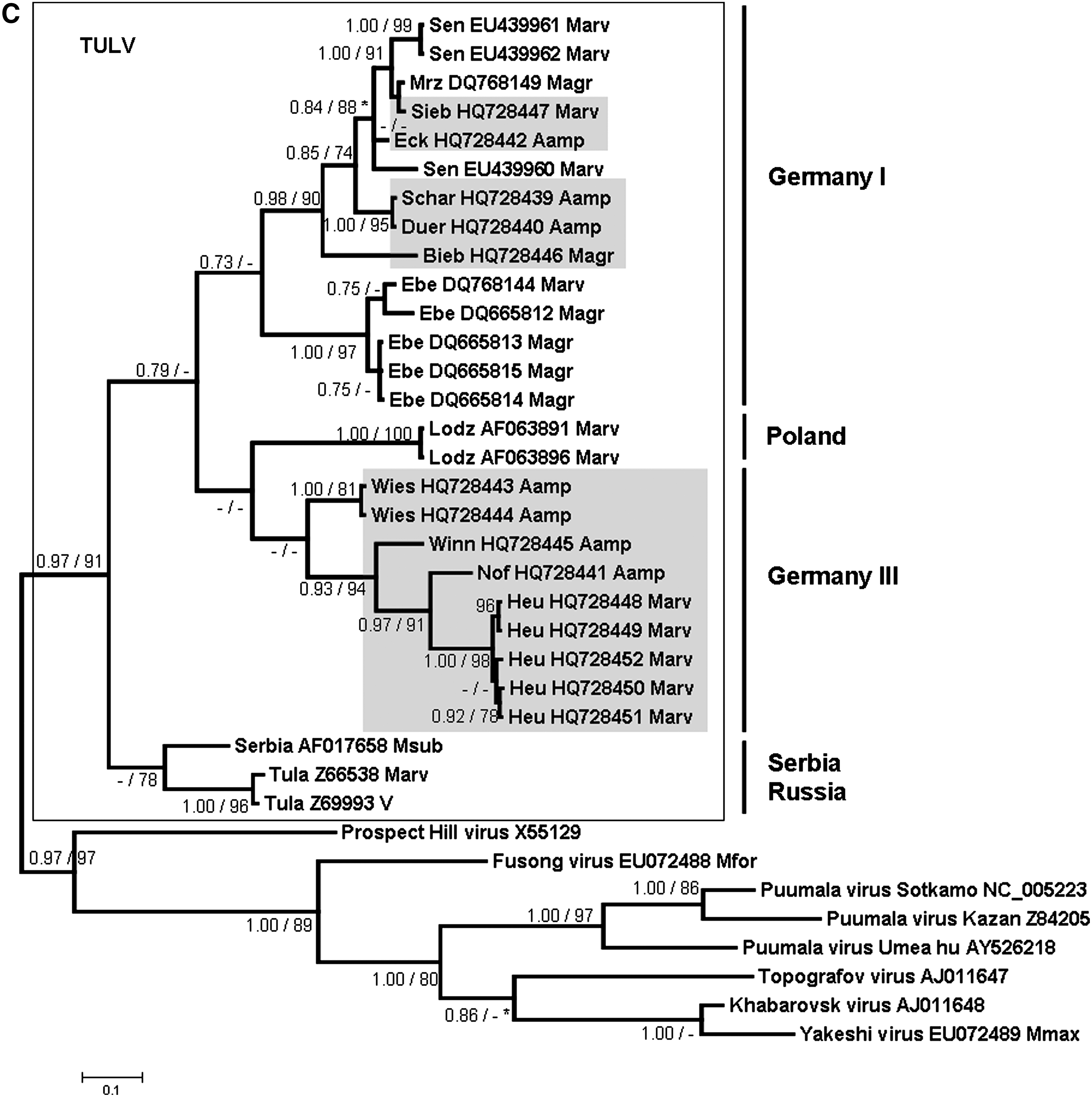
The phylogenetic analyses of these partial L-, S-, and M-segment sequences showed that the novel A. amphibius-derived sequences cluster geographically with TULV sequences derived from M. arvalis and M. agrestis in Germany and Europe, but are clearly separated from sequences of Microtus-borne Prospect Hill virus and Isla Vista virus and other Arvicolinae-derived hantaviruses (Fig. 2A, B, and C). All TULV S- and M-segment sequences from Arvicola are part of two well-supported groups “Germany I” and “Germany III/Switzerland.” For both segments, the first group contains sequences derived from A. amphibius from the federal states Thuringia and Saxony (trapping sites Eck, Schar, and Duer), clustering together with TULV sequences from M. arvalis (Marv) and M. agrestis (Magr) from Lower Saxony (trapping sites Sen and Goett), Thuringia (trapping site Sieb), and Brandenburg (trapping sites Bieb, Mrz, Ben, and Ebe) (Fig. 2B and C). The group “Germany III/Switzerland” comprises water vole-derived TULV sequences from Baden-Wuerttemberg (trapping sites Roem, Winn, and Wies), and Switzerland (trapping site Nof), as well as Microtus-derived sequences from Baden-Wuerttemberg (trapping site Heu), and Bavaria (trapping site Graf) (Fig. 2B and C). Pair-wise comparisons of the novel partial Arvicola-derived TULV S- and M-segment nucleotide sequences (and deduced amino acid sequences) showed divergences of 0.2–20.4% (0–8.4%) and 0.3–19.9% (0–1.6%), respectively (data not shown). The screening for recombination did not detect any putative recombinant regions.
Bayesian trees based on partial L-segment (336 nucleotides) (
The serological screening of 286 A. amphibius from 5 different trapping sites where CCF was available identified 16 reactive samples with prevalences of 2.3–27% (Fig. 1 and Table 1).
Phylogenetic analysis of cyt b sequences did not support evolutionary distinctness of the novel A. amphibius sequences from Germany and Switzerland, but rather showed relatively high similarity with other A. amphibius sequences from Switzerland and Finland (Fig. 3). The cyt b sequences derived from Spanish southern water voles A. sapidus formed an own cluster.
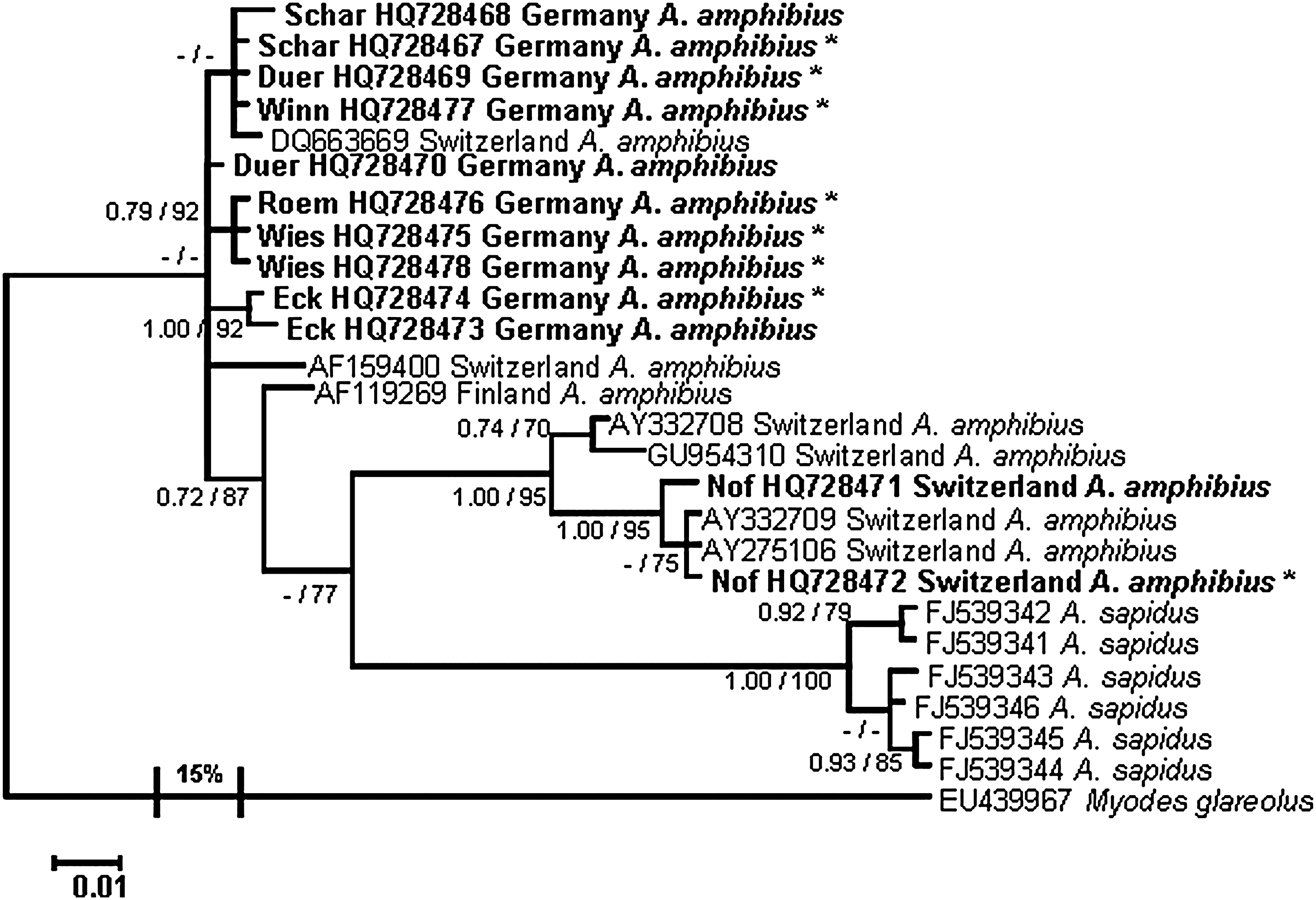
Bayesian tree based on partial cytochrome b sequences (608 bp) of Tula virus-RT-PCR-positive (*) and negative Arvicola amphibius from Germany and Switzerland. Posterior probabilities for Bayesian analysis are given before the slashes, and aLRT (approximate Likelihood-Ratio Test) values for branches for maximum likelihood analysis after the slashes. Only values ≥0.7% and ≥70% are shown; values ≤0.7% and ≤70% are indicated by hyphens. Scale bar indicates the number of nucleotide substitutions per site. The outgroup branch is condensed and the divergence to Arvicola spp. is given in percentages at the branches in the tree. The novel sequences are shown in bold. Sequences from the sister species A. sapidus, and additional sequences from A. amphibius available from GenBank were included, and their accession numbers are displayed in the tree. Myodes glareolus was used as an outgroup to root the phylogenetic tree.
Discussion
In this study we report the first multiple molecular evidence of Microtus-associated TULV infections in a representative of another genus of the subfamily Arvicolinae (i.e., in the Eurasian water vole). This finding is based on sequences from all three TULV genome segments from a large panel of water vole samples from different geographic regions in Germany and Switzerland. The observed frequent spillover (or host switch) is highly unexpected, as paleozoological and molecular investigations suggest a last common ancestor of Microtus and Arvicola more than three million years ago (Chaline and Graf 1988; Conroy and Cook 1999; Abramson et al. 2009; Fink et al. 2010). Such transmissions of a hantavirus from the original reservoir host to distantly related species other than humans have been reported only very rarely in Europe, as for example a single house mouse infected by Apodemus flavicollis-associated Dobrava-Belgrade virus (Weidmann et al. 2005). Spillover infections have been documented in numerous studies in the New World, with detection of Microtus-borne viruses in Peromyscus and Sigmodon species, Peromyscus-borne viruses in Reithrodontomys species, chipmunks and Mus species, Reithrodontomys-borne viruses in voles and wood rats, and Oryzomys-borne viruses in Sigmodon species (Ulrich et al. 2002).
The geographical clustering of the Arvicola-borne sequences with Microtus-derived TULV sequences from different parts of Germany and Europe suggests multiple spillover infections of TULV from M. arvalis or M. agrestis to A. amphibius. Although an opposite direction of the spillover infections seems possible, the frequency of molecular detection of TULV in Microtus species, however, suggests that members of this genus represent the reservoir host (Plyusnin et al. 1994; Sibold et al. 1995; Schmidt-Chanasit et al. 2010). In addition, the detection of TULV-reactive antibodies without detection of TULV-specific RNA in the water vole may also support this conclusion. Although the route of TULV transmission from Microtus to Arvicola is not known, both species are sometimes found in the same habitats and may even use the same burrows (G Heckel, R Wolf, personal communication).
Alternatively, the multiple molecular detection of TULV infections in water voles from several sites and different geographic regions in Germany and Switzerland indicates the potential of this rodent species as an additional reservoir host of this particular virus. Thus our findings also raise more general questions on the definition of a reservoir host for a given hantavirus and the role of hantavirus/rodent co-speciation in their molecular evolution. Usually the multiple detection of nucleic acid sequences in a single reservoir host and their absence in sympatrically-occurring other rodent or small mammal species is believed to be indicative of a reservoir host function (Hjelle and Yates 2001). Thus, M. arvalis has initially been identified as the most likely reservoir host of TULV (Plyusnin et al. 1994; Sibold et al. 1995). However, TULV was molecularly detected in additional Microtus species (i.e., M. levis, formerly M. rossiaemeridionalis), M. agrestis, M. subterraneus, and M. gregalis (Plyusnin et al. 1994; Sibold et al. 1995; Schmidt-Chanasit et al. 2010), and as here described in A. amphibius. Similarly, a TULV infection has been recorded in Lagurus lagurus, a representative from another genus of the Arvicolinae subfamily (GenBank accession numbers AF442619 and AF442618; Dekonenko and Yakimenko, unpublished data). Moreover, a previous study suggested an already established isolated replication and transmission cycle of TULV in M. agrestis (Schmidt-Chanasit et al. 2010). Finally, these findings are in contrast to the usually assumed co-evolution hypothesis, according to which one would expect different TULV lineages in Arvicola and Microtus reservoirs.
TULV has previously been detected in reservoir hosts from several parts of Germany and from Russia, Slovakia, Croatia, the Czech Republic, Austria, Poland, Belgium, France, Hungary, The Netherlands, and Slovenia (Schmidt-Chanasit et al. 2010). The herein described detection of TULV sequences in A. amphibius, M. arvalis, and M. agrestis from Thuringia and Baden-Wuerttemberg, southeast and southwest Germany, thus enlarges our knowledge of the geographical distribution of this virus. Obviously, this virus has a broad, likely Germany-wide, distribution. Moreover, the detection of a TULV sequence in Arvicola from Switzerland is the first molecular detection of this virus in Switzerland, as previously only a clinical case caused by TULV was reported, using focus reduction neutralization assay analysis (Schultze et al. 2002).
This broad geographical distribution of TULV in the reservoir host also raises questions about the reasons for the currently very low number of reported human TULV infections, with only two case reports of potential TULV-induced disease in humans (Schultze et al. 2002; Clement et al. 2003; Klempa et al. 2003). TULV-related Microtus-associated hantaviruses in the New World (i.e., Prospect Hill virus, Bloodland Lake virus, and Isla Vista virus) have not been shown to cause significant disease in humans (Lee et al. 1985; Hjelle et al. 1995; Song et al. 1995). Similarly, TULV and another Eurasian hantavirus species (Khabarovsk virus hosted by the reed vole Microtus fortis) (Hörling et al. 1996), are believed to have low or no pathogenicity for humans. The low frequency of the detection of human TULV infections might be explained by its low pathogenic potential as determined in cell culture experiments (Kraus et al. 2004). On the other hand, a recent study in a forestry worker risk group in a region where TULV has been detected in Microtus reservoirs demonstrated frequent serological detection of TULV-reactive antibodies in an ELISA test using the homologous antigen (Mertens et al. 2011). Therefore, more extended serological investigations should determine if TULV represents a neglected human pathogenic hantavirus that is currently overlooked in human infections due to the use of insufficient diagnostic tools.
In conclusion, multiple molecular detections of TULV infections in water voles underline the unique potential of this virus to infect distantly-related rodents of two different genera. Future studies of wild water voles and other Arvicola species from different regions in Europe are needed to verify the frequency of spillover and/or host-switch events for TULV, and may thus allow the definition of the host range of TULV. These studies should be accompanied by experimental infection studies addressing the pathogenic consequences of TULV infections in different hosts, and the viral and host factors determining the host range, transmission pathways, and human pathogenicity of this virus.
Footnotes
Acknowledgments
The authors kindly acknowledge the support of Dörte Kaufmann, Astrid Thomas, Kathrin Hirsbrunner, Cornelia Triebenbacher, Anja Globig, Gerhard Dobler, Daniel Windolph, Michael Noack, Paul-Walter Löhr, Johannes Lang, Thomas Schröder, Dietrich Heidecke, Jens Jacob, Hans-Joachim Pelz, Thorsten Menke, Hermann Ansorge, Denny Maaz, Matthias Tzschoppe, Lutz-Florian Otto, Martin Kaatz, as well as the helpful comments from Lutz C. Maul.
This work was financially supported by the German Federal Ministry of Food, Agriculture and Consumer Protection (BMELV), grant number 07HS027 (to R.G.U.).
Author Disclosure Statement
No competing financial interests exist.