
Abstract
A total of 821 tissue samples from rodents trapped during field campaigns organized in Europe and Africa were screened for the presence of arenaviruses by molecular methods and cell culture inoculation when feasible. Two Mus musculus domesticus trapped in the southwestern part of France were infected with a potentially new strain of lymphocytic choriomeningitis virus (LCMV), here referred to as LCMV strain HP65-2009, which was isolated and genetically characterized by whole genome sequencing. Genetic and phylogenetic analyses comparing LCMV HP65-2009 with 26 other LCMV strains showed that it represents a novel highly-divergent strain within the group of Mus musculus-associated LCMV.
Introduction
According to the International Classification for Taxonomy of Viruses (ICTV), the Arenaviridae currently includes 22 viruses classified in two monophyletic complexes (Old World [OW] and New World [NW]), on the basis of geographical, genetic, antigenic, and host relationships (Wulff 1978; Moncayo et al. 2001; Buchmeier 2007; Gonzalez et al. 2007). Four other viruses are listed as putative species: Kodoko, Dandenong, and Merino Walk viruses (Salvato et al. 2005), and Lujo virus (Briese et al. 2009). In addition, at least six newly described viruses—Gbagroube, Menekre (Coulibaly-N'golo et al. 2011), Catarina (Cajimat et al. 2007), Skinner tank (Cajimat et al. 2008), big brushy tank, and Tonto Creek (Milazzo et al. 2008)—are to be included in the family.
The LCMV prototype of the Arenaviridae family is the only member showing a worldwide distribution (most probably due to its association with the cosmopolitan rodent species Mus musculus), while all other arenaviruses seem to be restricted geographically (Charrel et al. 2008). LCMV is an OW arenavirus. Epidemiologic studies show that up to 11% of wild mice can be infected with LCMV (Lledo et al. 2003; Riera et al. 2005). Infected mice can shed the virus throughout their life. Humans are infected through inhalation of infectious rodent excreta or secreta, or by direct contact with infected rodents (Emonet et al. 2009). Of the OW arenaviruses species, only LCMV, Lassa, and the newly-discovered Lujo viruses are known to cause severe disease in humans (Buckley and Casals 1970; Briese et al. 2009). LCMV infection is typically asymptomatic or associated with mild transient illness. Yet the virus can sometimes cause aseptic meningitis and encephalitis (Martos Fernandez 1996; Barton and Hyndman 2000; De Ory et al. 2009), congenital malformations, or abortion (Barton and Mets 1999). Recently, fatal cases of infection have been reported after organ transplantation (Fischer 2006; Centers for Disease Control and Prevention 2008; Palacios et al. 2008).
We initiated a screening program targeting rodents for the presence of arenaviruses through an approach that combines molecular surveys using both specific and generic primers with cell culture inoculation. Here we provide results from individuals collected in the field between 2002 and 2009, in both Africa and Europe.
Materials and Methods
Study sites and rodent collection
Rodents were trapped during independent field trips conducted at different years and periods and in different countries, as detailed in Table 1. The rodents were sampled using Sherman and wire-mesh traps baited with flour and sardines (Europe), or peanut butter (Africa), set in the evening and checked in the morning, usually for several consecutive nights. When the determination was straightforward (e.g., Mus musculus, Desmodilliscus braueri, or Jaculus jaculus), the animals were identified to species level in the field on morphological grounds (Wilson 1993; Granjon et al. 2009). In the case of sibling species that may co-occur in the wild, such as for West African rodents, unambiguous species-specific identifications were performed by karyotyping and/or by a “cytochrome b-based bar-coding” approach (Dobigny and Yang 2008; Dobigny et al. 2011). During autopsy, one or more dissected organs (liver, spleen, lungs, heart, and kidneys) of each specimen were collected and stored individually at −80°C in liquid nitrogen, or in RNA-preserving buffers such as RNA Later (Sigma-Aldrich, St. Louis, MO) and RNA Now (Biogentex, League City, TX; Table 1).
Table presents the list of all rodents whose tissue samples were processed for arenaviruses, and also details about collection, storage, primer sets, and screening results. The Numbers besides the species indicate the quantity of captured animals. Details about rodents from which viruses were isolated in our are indicated with bold type.
LCMV, lymphocytic choriomeningitis virus.
Processing rodent tissues
Frozen samples were thawed and ground mechanically with a 3-mm tungsten carbide bead using a Mixer Mill MM300 (Qiagen SA, Courtaboeuf, France) at 24 cycles/sec for 5 min, with 600 μL of minimal essential medium (MEM) supplemented with 3% fetal bovine serum (FBS), 1% glutamine, 1% penicillin, 1% streptomycin, and 3% kanamycin. Homogenized tissues were centrifuged at 13,000 g for 5 min. An aliquot of 200 μL of the supernatant was prepared and used for viral nucleic acid purification with the EZ1 virus mini kit v 2.0 on the BioRobot EZ1XL (both from Qiagen SA), to a final elution volume of 60 μL. Further analyses were performed using pools of samples prepared by mixing homogenates from 8–10 samples of tissues of the same organ type, and of rodents coming from the same locality. When positive results were obtained, tests were repeated for every sample constituting the pool.
Cell culture inoculation
All work with infectious material was performed in a Biosafety Level-3 (BSL-3) laboratory in Marseille, France. An aliquot of 100 μL of the clarified supernatant was used to inoculate Vero cells in 12.5-cm2 plastic tissue flasks. The cells were maintained in MEM supplemented as described above and incubated at 37°C. The flasks were checked daily using an inverted microscope for cytopathic effect (CPE). On day 8 post-inoculation, the cell culture fluid overlay was discarded and total nucleic acids were purified.
Detection of arenaviruses by RT-PCR using generic and specific assays
Investigations for the RT-PCR detection of arenaviruses were conducted on nucleic acids obtained directly from tissue samples, and from supernatants of inoculated cell cultures. Three different conventional PCR assays were used.
The first two systems targeted the same region in the NP gene; one was generic for all arenaviruses, whereas the second was specific to LCMV. The first system used the primers 1010C and 1696R (Bowen et al. 2000). The second system (nested RT-PCR) used primers 1817V-LCM and 2477C-LCM for the first round, and primers 1902V-LCM and 2346C-LCM for the second round (Emonet et al. 2007). First round RT-PCR used the One-Step Access RT-PCR kit (Promega Corp., Madison, WI), according to the manufacturer's protocol, with a hybridization step at 55°C for 1 min. Nested PCRs were performed using the same conditions with 1.25 U of Taq DNA polymerase (Invitrogen, Carlsbad, CA).
The third system targeted the L gene of OW arenaviruses, and used primers LVL3359A-plus, LVL3359D-plus, LVL3359G-plus, LVL3754A-minus, and LVL3754D-minus (Vieth et al. 2007). RT-PCR was also performed using the One-Step Access RT-PCR kit (Promega Corp.) according to the manufacturer's protocol, with a hybridization step at 55°C for 1 min. PCR products were loaded on 1.5% agarose gels, and stained with ethidium bromide. Amplification products of the expected size were purified with the QIAquick PCR purification kit (Qiagen), and directly sequenced using the BigDye® Terminator v. 3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA).
Sequence determination, molecular characterization, and phylogenetic analysis
Viral RNA obtained from infected Vero cells was used to extend sequences obtained at the detection stage. RT-PCR was performed using 5 μL of viral RNA with the One-Step Access RT-PCR kit. Primers used for sequencing are presented in Supplementary Table 3 (see online supplementary material at
Phylogenetic studies were performed using MEGA v.5.0 (Tamura et al. 2011) and Geneious Pro™ v.5.4.6 (Drummond 2011). Genetic distances at the amino acid (AA) and nucleotide levels were calculated with the p-distance method. Phylogenetic trees were computed using: (1) the neighbor-joining (NJ) method based on the p-distance substitution model, (2) the maximum likelihood (ML) method based on the Jones, Taylor, and Thornton (JTT) matrix-based model, and (3) the maximum parsimony (MP) method based on the Close-Neighbor-Interchange with exclusion of parsimony non-informative sites from the alignment. The robustness of the nodes was tested by 10,000 bootstrap pseudoreplications.
To check the presence of either a recombination or a reassortment event within sequences of the novel LCMV strain, a control test was performed using the Recombination Analysis Tool (RAT; Etherington et al. 2005).
Results
Rodent sampling and arenavirus detection
A total of 821 rodents representing 18 different genera were trapped in Africa and Europe (Table 1). Heart, liver, lungs, spleen, and kidney tissue samples were collected and stored either at −80°C using liquid nitrogen, or at −20°C in RNAlater. Of all screened rodent tissue samples, only 289 were stored at −80°C, which allowed detection through virus culture.
None of the 156 Mastomys (including 89 M. natalensis, the species known as the main reservoir for Lassa fever virus; Lecompte et al. 2006) were found positive for Lassa virus or another arenavirus.
Of the seven Mus musculus captured in a dog kernel in the French area of Hautes-Pyrénées in 2009, two were positive with the second and the third of the three conventional systems targeting all arenaviruses. The tested specimen consisted of pools of different organs, all gathered from the same animal. Searches using BLASTN and BLASTX indicated that these four PCR products (two from each animal) had high similarities (∼80%) with sequences related to LCMV, but were distinct from homologous sequences retrieved from the GenBank database. The strains were named HP65-2009/1 and HP65-2009/2, with “HP” standing for the name and “65” for the number of the area where the infected mice were captured.
Virus isolation, electron microscopy, and antigenic characterization
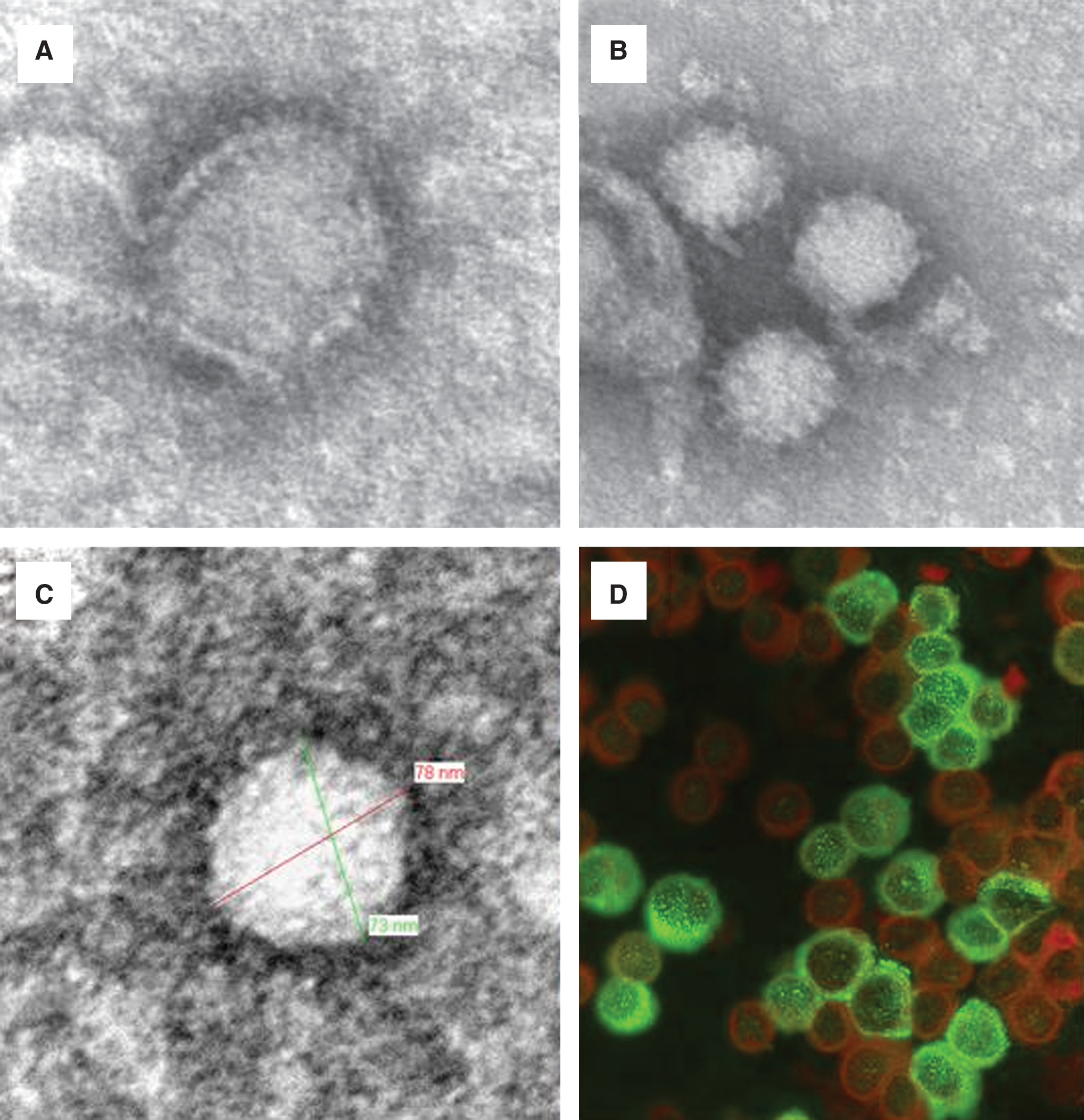
Both the HP65-2009/1 and HP65-2009/2 strains were isolated by cultivation in monolayer cultures of Vero E6 cells. The supernatant was prepared for electron microscopy (EM) analysis. EM micrographs (Fig. 1A, B, and C) showed viral particles with a morphology characteristic of arenaviruses (Dalton et al. 1968; Murphy and Whitfield 1975). In Figure 1D, Vero cells infected with the LCMV HP65-2009/1 strain are shown reacting in an indirect immunofluorescence assay (Charrel et al. 2009) with the serum of a convalescent patient who experienced past infection with the Marseille strain of LCMV (Emonet et al. 2007), thus demonstrating the expected cross-reactivity.
Full-length sequence of LCMV strain HP65-2009/1 and comparative genetic analysis
The supernatant from a culture of Vero cells infected with LCMV HP65-2009/1 strain was used to determine the complete sequence of the S and L RNA segments. Full-length sequences of the 2 RNA segments of LCMV HP65-2009/1 were registered in the Genbank database with accession numbers JF912084 and JF912085 for the S and L RNA segments, respectively. Using the supernatant from the culture of Vero cells infected with LCMV strain HP65-2009/2, four partial nucleotide sequences of the LCMV strain HP65-2009/2 were also determined, corresponding to the NP, GPC, L, and Z genes. These four sequences were registered in the Genbank database with accession numbers JN546573, JN546574, JN546575, and JN546576, for the Z, L, GPC, and NP genes, respectively.
Genetic analysis of untranslated regions
The 19-nucleotide (nt)-long sequences located at the extreme 3′ and 5′ ends of LCMV strain HP65-2009/1 were inferred from the sequence of oligonucleotide ARE-END, in view of the fact that these regions are reverse complementary to each other, are highly conserved in other arenaviruses (Meyer et al. 2002), and that ARE-END primed for cDNA synthesis and PCR amplification. The 5′ and 3′ non-coding regions (NCRs) of the S segment are 66- and 61-nt long, respectively. The S segment intergenic region (IR) is 70-nt in length (positions 1561–1624). The predicted secondary structure of the LCMV HP65-2009/1 IR includes a single stem-loop structure (Auperin et al. 1984; Romanowski and Bishop 1985; Auperin et al. 1986; Charrel et al. 2001; Moncayo et al. 2001; Archer and Rico-Hesse 2002). The L segment has a 5′ NCR of 89 nt, a 3′ NCR of 32 nt, and an IR of 210 nt, which is assumed to form a complex folding structure, as observed in other arenavirus L segment IRs (Archer and Rico-Hesse 2002; Briese et al. 2009).
Genetic analysis of coding regions
The S RNA of LCMV strain HP65-2009/1 is 3366 nt long. The virus GPC is encoded in an open-reading frame (ORF) that is 1494 nt long, with the initiation codon located at nucleotides 67–69, and a translation termination signal UAA at nucleotides 1561–1563. The length of the LCMV HP65-2009/1 GPC gene ORF (498 AA) is similar to that of other known LCMV strains. The GPC sequence diverges from that of other known LCMV strains by 8–21% and by 18–35% at the amino-acid and nucleotide levels, respectively. The NP gene comprises an ORF that is 1674 nt long, with the start codon at nucleotides 3304–3302 (nt positions numbered from the 5′ end of the S RNA), and a UAA stop codon at nucleotides 1630–1628. The length of the NP gene (558 AA) is similar to that of all other existing LCMV strains. The sequence differs from that of other LCMV strains by 7–14% and by 17–27% at the AA and nucleotide levels, respectively.
The L RNA is 7228 nt long. The polymerase gene comprises an ORF that is 6627 nt long with a start codon located at nucleotides 7196–7194 (nt positions numbered from the 5′ end of the L RNA), and a UGA termination codon at nucleotides 569–567. The length of the polymerase gene (2209 AA) is similar to that of all other existing LCMV strains. The sequence diverges from that of other LCMV strains by 12–21% and by 25–33% at the AA and nucleotide levels, respectively. The Z gene is encoded in an ORF that is 270 nt long, with the start codon located at nucleotides 90–92, and a UGA stop codon at nt 360–362. The length of the Z gene (90 AA) is similar to that of other LCMV strains. The sequence differs from that of other LCMV strains by 10–21% at the AA level and by 11–19% at the nucleotide level.
The sequences determined in our study from both house mice were most closely related to each other than to any other LCMV sequences: a comparison of partial sequences of the S RNA of HP65-2009/1 and HP65-2009/2 showed respective divergences of 3.05% from nt 332–658 (in the GPC gene), and of 0.61% from nt 1707–2284 (in the NP gene); similar comparisons in the L RNA showed respective divergences of 2.90% from nt 2–435 (in the Z gene), and of 1.28% from nt 3674–3993 (in the L gene).
Pairwise distances between LCMV strains and selected Old World arenaviruses were computed for nucleotides as well as for amino acid alignments (Supplementary Tables 2a, 2b, 2c, and 2d; see online supplementary material at
Phylogenetic analysis
Results of various phylogenetic analyses performed using full-length S- and L-segment sequences are shown in Figure 2. Complete homologous genomic data were available for a total of 27 LCMV strains (Supplementary Table 1; see online supplementary material at
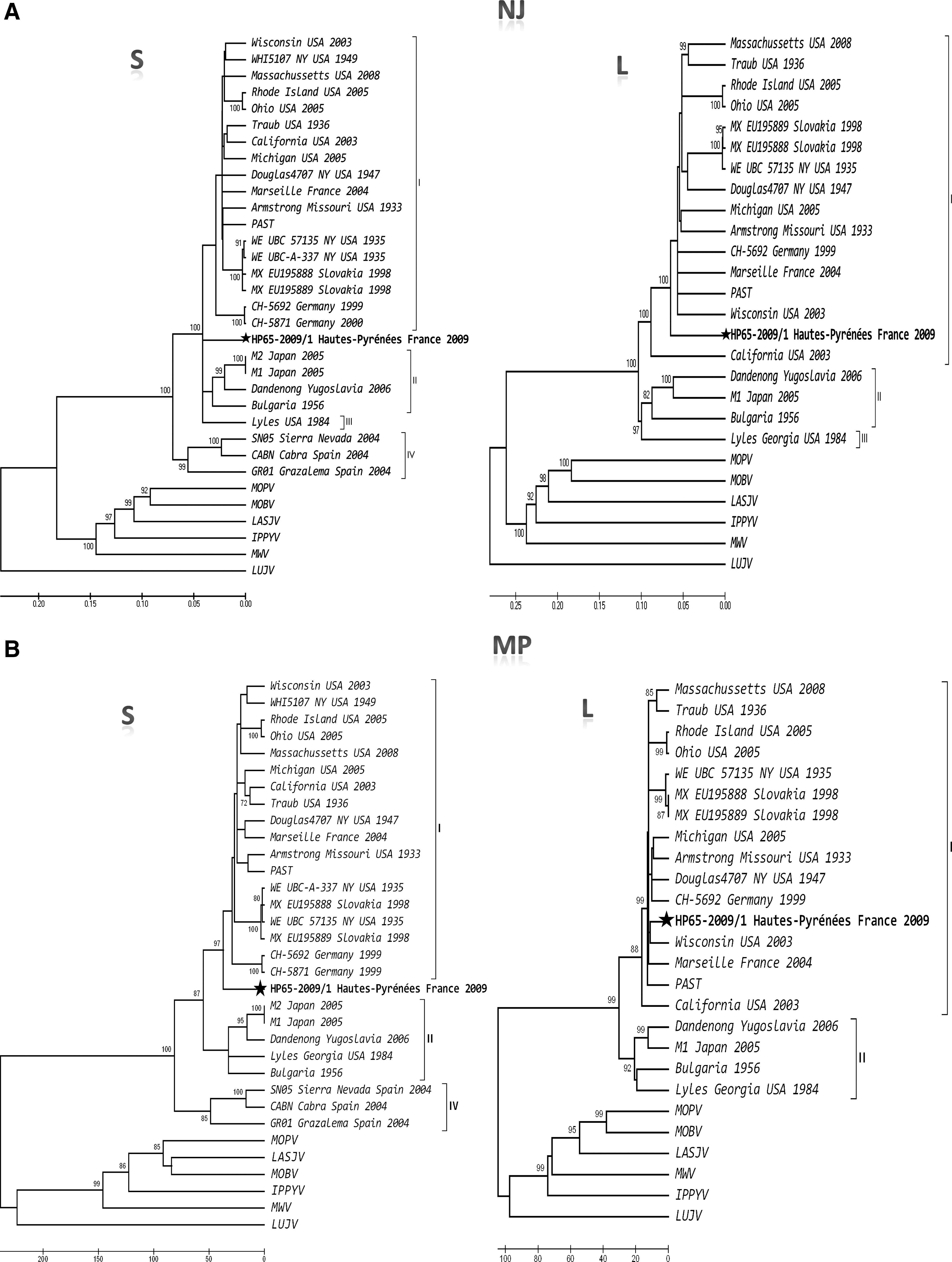
Phylogeny of Old World arenaviruses and the virus detected in this study based on the analysis of full-length S- and L-segment sequences. The evolutionary history was inferred using the neighbor-joining (

Discussion
The aim of this work was to investigate the presence of known or potentially novel arenaviruses in rodents trapped during field campaigns in different countries of Europe and Africa, to isolate the corresponding strains and to characterize them through complete genome sequencing and phylogenetic analyses.
In spite of a reasonable sampling effort, no arenavirus could be detected in any of the 379 African rodents investigated here, or in the 358 rodents trapped in Germany, Portugal, and Slovakia.
On the contrary, only 80 rodents were scrutinized in France, and evidence for arenaviruses was observed in two Mus musculus domesticus animals trapped in the southwestern part of the country. Sequence analyses allowed us to unambiguously identify LCMV. Genetic and phylogenetic analyses performed with the AA sequences of both S and L RNA segments showed that (1) all LCMV strains were consistently grouped together, with bootstrap values reaching 100%, which represents strong evidence of a common ancestry for all LCMV strains, and confirms previous findings (Gallaher et al. 2001; Albarino et al. 2010); (2) LCMV strains showed high levels of genetic diversity, which are comparable to those observed in Lassa virus (Bowen et al. 2000; Gunther and Lenz 2004); (3) LCMV strains associated with Mus musculus on one hand, and LCMV strains isolated from Apodemus sylvaticus on the other hand, represent distinct clades; (4) the LCMV HP65-2009/1 strain was consistently grouped with LCMV strains later isolated from and associated with Mus musculus; (5) and although LCMV HP65-2009/1 is one of the most divergent of all currently known Mus musculus-associated LCMV strains, it undoubtedly belongs to the species LCMV.
Moreover, the discordance observed in the positioning of the LCMV HP65-2009/1 strain between the S- and L-segment-based phylograms, computed using different reconstruction methods, raises the hypothesis of a possible reassortment within the S RNA segment of the novel strain. Yet the recombination test did not confirm this hypothesis. Therefore, although the novel HP65-2009/1 strain is genetically one of the most divergent among all LCMV strains, it appears to belong to lineage I. Nevertheless, our findings of a new genetically divergent strain suggest that the diversity within LCMV species may be much higher than initially perceived. Future discoveries of new strains may confirm this point, and if true, may require us to revisit the genetic criteria ruling the definition of species in the genus Arenavirus.
Successive passages of viruses during cell culture can lead to sequence mutations or virus pathogenicity attenuation as a consequence of a possible adaptation of the viruses to the cells (Chen et al. 2003; Song et al. 2003; Anez et al. 2009; Chambers et al. 2009; Fu et al. 2010). In our case, a comparison of partial sequences of both strains obtained from the tissue samples with those sequenced using the cell supernatant did not show any nucleotide difference.
The main reservoir of LCMV is Mus spp. (Ackermann et al. 1964; Childs et al. 1992; Morita et al. 1996). Our discovery of potentially new strains of LCMV in Mus musculus reinforces this notion. However, the discovery of the Spanish LCMV GR01, CABN, and SN05 strains in Apodemus sylvaticus (Ledesma et al. 2009) suggested that this species could be an alternative reservoir. In other studies LCMV antibodies were detected in Clethrionomys glareolus, Apodemus flavicollis, Microtus arvalis, and Apodemus sylvaticus (Kallio-Kokko et al. 2006), Neotoma spp. (Kosoy et al. 1996), and Rattus norvegicus (Ledesma et al. 2009). There have been reports of LCMV infections of humans after close contact with hamsters (Rousseau et al. 1997; Amman et al. 2007), and also of interest are the recent reports of human-to-human transmission via organ transplantation (Fischer et al. 2006; Razonable 2011). Altogether, these data suggest that the host diversity could be more important than previously perceived, hence the importance of re-examining the diversity of LCMV and arenavirus reservoirs.
As previously reported (Albarino et al. 2010), there is an absence of correlation between phylogenetic position and collection date of the virus. This fact is intriguing, since this is unique to the genus Arenavirus. Indeed, similar studies performed with other arenavirus species (Lassa, Junin, Guanarito, Pirital, and Whitewater Arroyo) showed well-resolved intra-species relationships with a gradual genetic differentiation among geographic isolates (Bowen et al. 2000; Garcia et al. 2000; Weaver et al. 2000; Fulhorst et al. 2001; Weaver et al. 2001). For instance, Lassa virus topology reflected a geographic clustering that parallels an east-to-west gradient (Bowen et al. 2000; Gunther et al. 2000). Similar findings were reported for Whitewater Arroyo virus in the southwestern United States, with a clear genetic grouping mirroring the geographic distribution (Fulhorst et al. 2001). The Pirital, Guanarito, and Junin viruses were also studied, but in smaller geographic areas; the correlation between genetic and geographic distances was less obvious, probably due to the smaller geographic range. In all cases, a dominant genotype was found surrounded by many other genotypes with restricted localization (Garcia et al. 2000; Weaver et al. 2000, 2001). If geographic distance seems to play a significant role in the discrimination of isolates of arenavirus species other than LCMV, neither the date of isolation nor the pathogenicity of the isolates could be associated with the genetic distance (Bowen et al. 2000; Garcia et al. 2000). Accordingly, data obtained for five arenaviruses (Lassa, Pirital, Guanarito, Junin, and Whitewater Arroyo) suggest that arenavirus evolution mainly follows a “parapatric-like” model: a continuously distributed population of dominant quasi-species, with gradual genetic separation linked to the history of rodent migration.
The apparent discordance observed here with LCMV between geographic and genetic distances could be explained by the nature of the reservoir host rodent, the house mouse, that has reached a worldwide distribution due to its close association with human activities (Boursot et al. 1993). As such, the lack of a correlation between phylogenetic position and date of virus isolation is consistent with a long and complex evolutionary history involving the frequent movement of rodent reservoir hosts that has accelerated during the last 500 years (Albarino et al. 2010). The genetic gaps observed here between Mus-associated and Apodemus-associated viruses also points toward the probable existence of still unknown LCMV-like viruses that remain to be discovered, not only in mice but also in other rodents.
Footnotes
Acknowledgments
Ines Yama was funded by the Fondation de Coopération Scientifique “Infectiopole Sud.” We are grateful to Maxime Galan, Madougou Garba, Philippe Gauthier, Jean-Grégoire Kayoum, Karmadine Hima, Abdoullaye Oumarou, Taiana Rivière-Dobigny, Bodo Seyni, and Caroline Tatard for their contribution in the field. Research permit in Niger no. 0648/MESS/R/T/SG/DRS/IT was provided to G.D. by the Direction de la Recherche Scientifique et de l'Innovation Technologique, Niamey, Niger.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.