
Abstract
The current work focuses on the study of polymeric, biodegradable nanoparticles (NPs) for the encapsulation of doxorubicin and mitomycin C (anti-leishmanial drugs), and their efficient delivery to macrophages, the parasite's home. The biodegradable polymer methoxypoly-(ethylene glycol)-b-poly (lactic acid) (MPEG-PLA) was used to prepare polymeric NPs encapsulating doxorubicin and mitomycin C. The morphology, mean diameter, and surface area of spherical NPs were determined by transmission electron microscopy (TEM), field emission scanning electron microscopy (FESEM), and BET surface area analysis. X-ray diffraction was performed to validate drug encapsulation. An in vitro release profile of the drugs suggested a fairly slow release. These polymeric NPs were efficiently capable of releasing drug inside macrophages at a slower pace than the free drug, which was monitored by epi-fluorescence microscopy. Encapsulation of doxorubicin and mitomycin C into NPs also decreases cellular toxicity in mouse macrophages (J774.1A).
Introduction
The Leishmania parasite exists in two life forms: an elongated, flagellated promastigote in the midgut of the sandfly, and small, rounded and non-motile amastigotes in macrophages. Macrophages engulf promastigotes via phagocytosis inside phagosomes, and these phagosomes fuse with lysosomes to form phagolysosomes, where they are converted to amastigotes (Gupta et al. 2010). Leishmania is the only known parasite that replicates inside macrophages and resists its own killing by an unknown mechanism (Ofec et al. 1995). Macrophages are the primary cells that invade different kinds of pathogens, and act as the first line of defense by phagocytosis of foreign particles (Gordon 1995). This strategy of macrophages can be used to actively deliver a drug directly to the site of infection, thereby reducing toxicity of the drug. Nano-based drug delivery systems have gained enormous attention due to their high stability, low toxicity, and tunable hydrophilic-hydrophobic behavior (Frank et al. 2008). Recently, efforts have been made to develop drug carriers such as peptide-conjugates, micelles, and liposomes (Jones and Leroux 1995; Torchilinn and Trubetskoy 1995; Panyam and Labhasetwar 2003). Some of these formulations are easily identified by the reticuloendothelial system, and are taken up by macrophages much more efficiently than free drug (Bally et al. 1998; Lundberg et al. 2004). It improves the efficacy of the drug, and greatly decreases its toxicity. Liposomal doxorubicin has reduced toxic effects without losing its efficacy against cancer (Tardi et al. 1996). A major drawback of liposomal formulations is their large size and extremely hydrophobic behavior, which leads to their accumulation at the injection site and poor solubility.
Recently, biodegradable NPs have been used extensively for the clinical administration of several anticancer drugs because of their rapid extravasation into tumor sites and controlled delivery (Desai et al. 1997; Brigger et al. 2002; Service 2005). Poly-(D,L-lactide) (PLA), a synthetic polymer, is hydrolyzed to nontoxic hydroxyl-carboxylic acid through ester bond cleavage, which is finally converted to water and carbon dioxide through the citric acid cycle. PLA has been approved by the U.S. Food and Drug Administration (FDA) due to its biodegradability (Xiao et al. 2010). PLA NPs exhibited an enhancement of the anti-leishmanial effects of primaquine (Muriel et al. 1998). MPEG-PLA diblock copolymer has been used for intravenous drug delivery due to its biodegradability and non-toxic behavior (Gref et al. 1995).
In this study, we intended to formulate these anti-leishmanial drugs encapsulated inside the polymeric nanospheres of MPEG-PLA copolymer to take advantage of their decreased toxicity, enhanced and localized delivery, slow release at the infection site, and increased efficiency. We chose low-molecular-weight hydrophilic MPEG (550) and a large amount of hydrophobic PLA to increase the overall hydrophobicity of the NPs. This increases absorption of plasma proteins and opsonins on the NP surface, thus facilitating phagocytosis by macrophages. The PLA segment of the polymer is hydrophobic and forms the inner core to entrap the doxorubicin and mitomycin C. We have successfully encapsulated doxorubicin and mitomycin C inside these polymer NPs, compared their toxicity level with free drug, and optimized delivery of the drug inside macrophages. Thus a logical strategy was developed for the efficient delivery of anti-leishmanial drugs using biodegradable polymeric NPs as drug carriers.
Materials and Methods
Chemicals
Doxorubicin, mitomycin C, MPEG (MW 550), PLA, stannous octoate [Sn(Oct)2], dimethyl sulfoxide (DMSO), dichloromethane (DCM), and acetonitrile, were purchased from Sigma-Aldrich (St. Louis, MO). DMEM culture medium, fetal bovine serum (FBS), and a penicillin-streptomycin antibiotic mixture were procured from Gibco (Invitrogen, Carlsbad, CA).
Maintenance of the cell line
Mouse macrophages (J774A.1) were procured from the National Centre for Cell Science, Pune, India. The cells were grown in DMEM containing 1.5 g/L NaHCO3, 10% FBS, penicillin (100 U/mL), and streptomycin (100 U/mL) at 37°C in 5% CO2.
Synthesis of the MPEG-PLA block copolymer
The MPEG-PLA diblock biodegradable co-polymer was synthesized using the ring opening polymerization method (Cheng et al. 2007; Zheng et al. 2010). In brief, in a three-necked round-bottom flask, 4 g lactide (3,6-dimethyl-1,4-dioxane-2,5-dione), and 10 g low-molecular-weight MPEG (550) were mixed in 30 mL distilled toluene (distilled by azeotropic distillation), containing 0.75% Sn(Oct)2 under a N2 flame. The flask was sealed and maintained at 130°C for 24 h in a silicon-oil bath. The synthesized polymer was dissolved in DCM and recovered by fractional precipitation using cold methanol. The final co-polymer was filtered and dried at 40°C in a vacuum for 48 h. The synthesized polymer was characterized by 1H-NMR (Varian) and FT–IR using a PerkinElmer spectrophotometer (model: Spectrum).
Formulation of drug-encapsulated nanoparticles
The nanoprecipitation method was employed for the synthesis of drug-encapsulated MPEG-PLA NPs (Fessi et al. 1989; Dong and Feng 2004). In brief, 1 mg of drugs (doxorubicin and mitomycin C) and 100 mg of MPEG-PLA block copolymer were dissolved in 10 mL of acetonitrile kept at 50°C to form the organic phase. This organic solution was added drop-wise to deionized water with 0.25% Pluronic F-68 as surfactant under moderate mechanical stirring. After 10 min, the MPEG-PLA block copolymer self-assembled into NPs due to diffusion of acetonitrile into the water with encapsulation of drug within its hydrophobic core. The acetonitrile was evaporated overnight at room temperature. Blank MPEG-PLA NPs were prepared using the same method but without the drugs.
Characterization of the synthesized nanoparticles
The formulated nanocapsules were characterized with respect to their morphology, size, surface area, and in vitro drug release properties.
Morphology and size determination
The morphology and size of the NPs were determined using field emission scanning electron microscopy (FESEM), and transmission electron microscopy (TEM). A drop of NP suspension, kept on aluminium foil, was dried at 60°C in an oven and attached to copper tape. After gold coating, the NPs were analyzed under FESEM using a Sigma Carl-Zeiss microscope operating at 1.3 kV EHT and 59.99 k× magnification. For the TEM exam, the sample was coated onto a carbon-coated copper TEM grid and air-dried. The sample was analyzed by a high-resolution TEM (JEM 2100; Jeol, Peabody, MA) operating at an accelerating voltage of 200 KeV.
Surface area, pore volume, and pore diameter of the NPs were determined with a BET surface area analyzer (Beckman Coulter, Brea, CA). Dried NPs (50 mg) were analyzed by BET at an outgas temperature of 150°C under N2.
X-ray diffraction and infrared spectroscopy studies
Determination of crystallographic properties and drug encapsulation of NP were done using an x-ray diffractometer. MPEG-PLA, MPEG-PLA doxorubicin NP, and doxorubicin were dried and analyzed by x-ray powder diffraction (XRD) using a Bruker D8 Advance x-ray diffractometer with Cu Kα radiation.
In vitro drug release
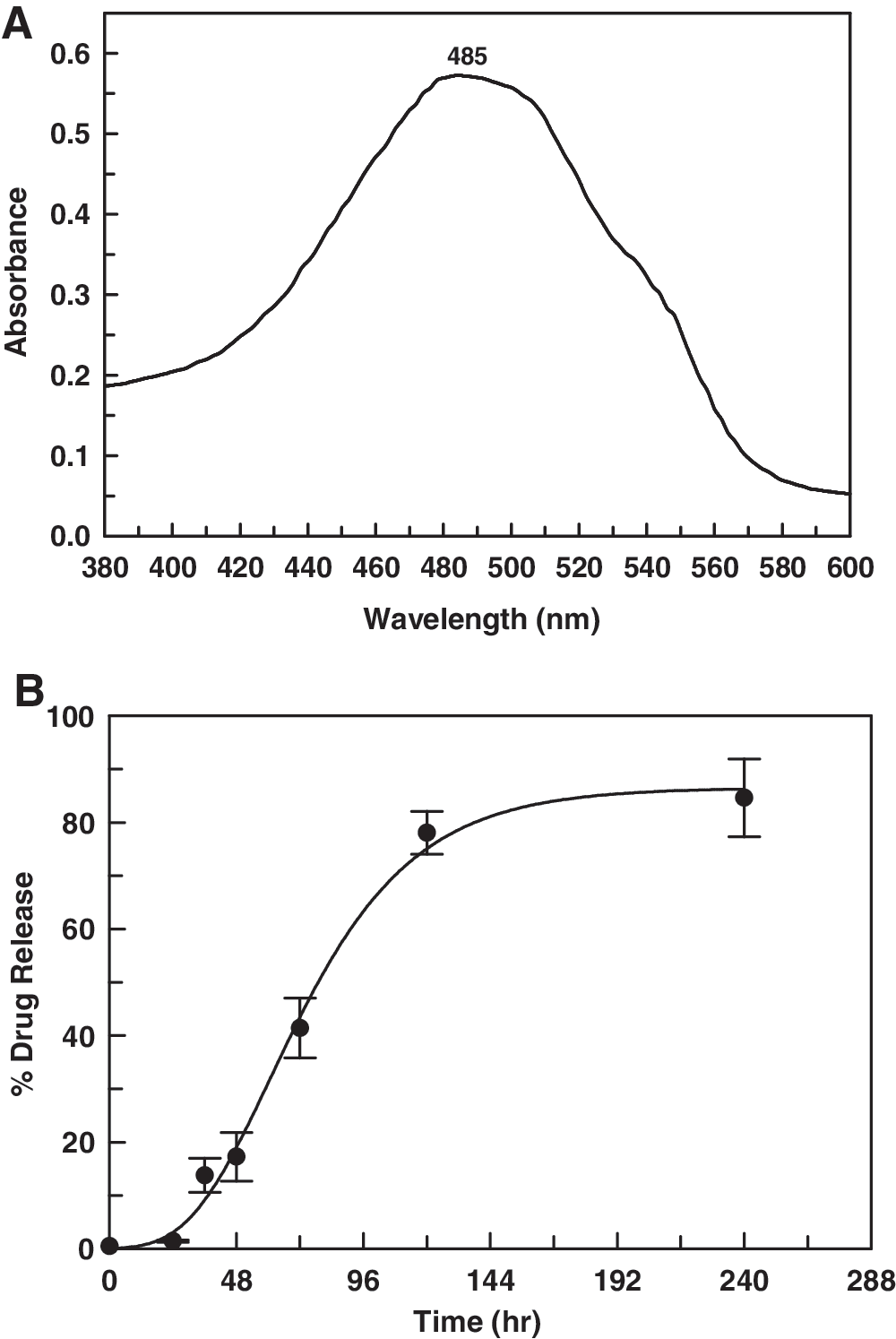
For determination of in vitro release kinetics of drug from MPEG-PLA NPs, 2 mL of doxo-NP solution was diluted to 20 mL with phosphate-buffered saline (PBS; pH 7.4), and kept in an orbital incubator shaker at 37°C and 30 rpm. The tube was taken out at different time intervals, centrifuged at 6000 rpm to pellet out NPs, and the supernatant was analyzed for drug content with a UV-VIS spectrophotometer at 485 nm. Doxorubicin showed absorbance maxima at 485 nm.
Optimization of macrophage release of encapsulated drug
Mouse macrophage (J774A.1) cells (1×106/mL) were seeded overnight in 24-well plates in DMEM with 10% FBS. MPEG-PLA-encapsulated doxorubicin and free doxorubicin were added to the culture at a final concentration of 10 μM. Fluorescence images of drug-loaded macrophages were collected to evaluate doxorubicin concentration inside the macrophages. The media was taken out at different time intervals (0, 8, 24, 48, 72, and 96 h), the cells were washed twice with PBS (pH 7.4), and analyzed under an epi-fluorescence microscope. Doxorubicin has red fluorescence when excited at 488 nm with emission maxima at 595 nm. Fluorescence intensity measurements were also performed using a Tecan (Frederick, MD) spectrophotometer (excitation 488 nm, emission 595 nm).
Cytotoxicity studies
The percentage cell viability was estimated on J774.1A mouse macrophages using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Mosmann 1983). Cells (1×106 cells/mL) were incubated in presence of different concentrations of control doxorubicin, mitomycin C, MPEG-PLA-doxo, and MPEG-PLA-mito (0–50 μM) for 24 h. After incubation with MTT for 4 h, a purple formazan complex formed, which was dissolved in DMSO and estimated at 570 nm. Viability of control cells without drugs was considered to be 100%. The experiment was performed in triplicate and the results presented are the mean values. The standard deviation is represented as error bars in the graph.
Results
Synthesis and characterization of the MPEG-PLA diblock co-polymer
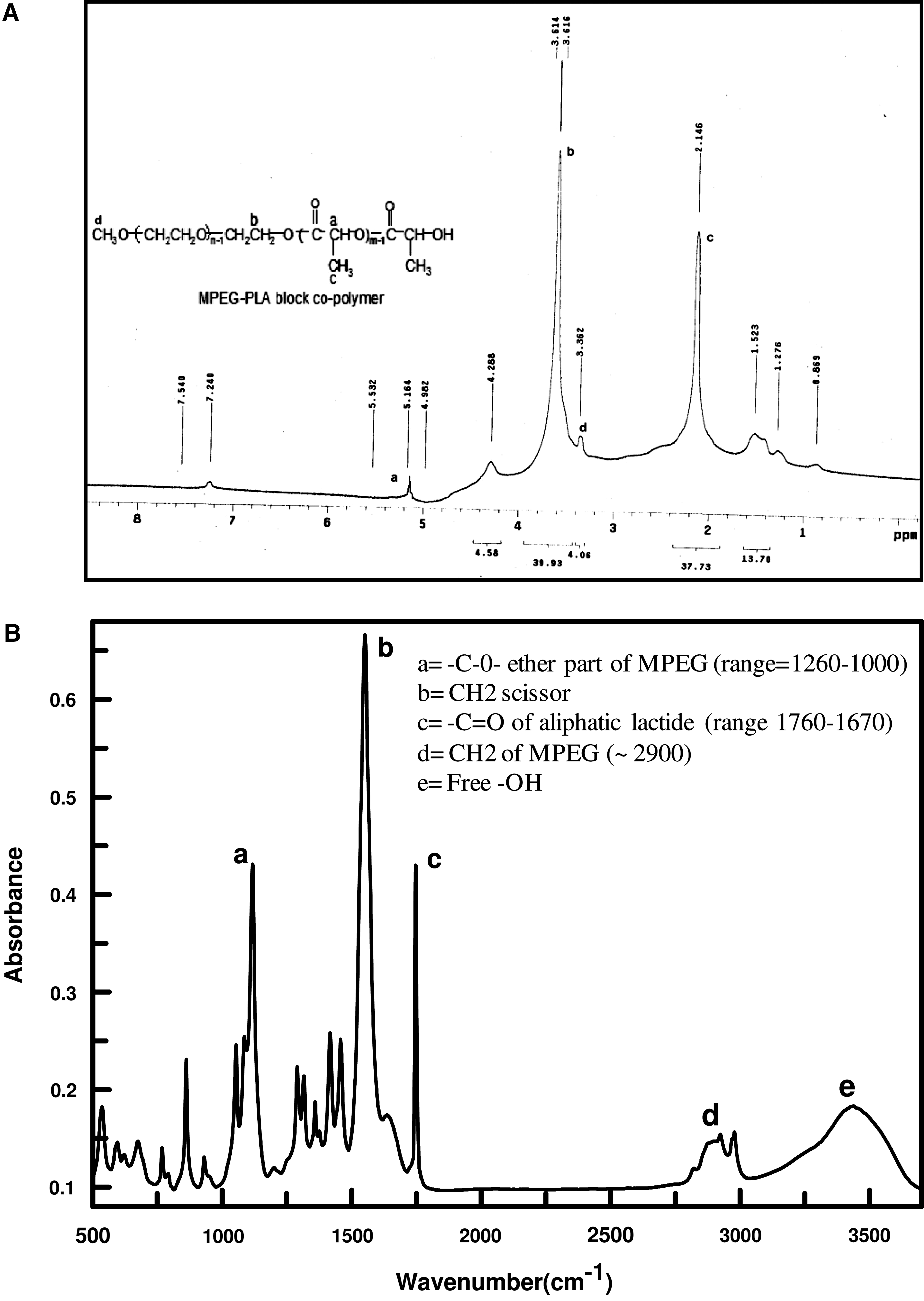
The MPEG-PLA diblock co-polymer was synthesized using MPEG and lactide as substrates, and highly efficient stannous octoate as a catalyst. The hydroxyl group (−OH) of MPEG interacts with the carboxyl (−COOH) group of opened lactide ring, which leads to the formation of an MPEG-PLA amphiphilic diblock copolymer (MPEG=hydrophilic core, PLA=hydrophobic core). The structure of the synthesized polymer was determined by 1H-NMR, taking CDCl3 as standard, and FTIR spectroscopy (Fig. 1). Peaks at 3.36 and 3.61 ppm correspond to ethereal CH3 and CH2 groups of MPEG, whereas peaks at 2.14 and 5.16 ppm correspond to the CH3 and CH groups of PLA in H1-NMR (Fig. 1A). FT-IR spectra also showed peaks corresponding to different functional groups of MPEG-PLA copolymer (i.e., peaks a and d correspond to the −O- [ether] and >CH2 group of MPEG; c, b, and e represent >CO of lactide, >CH2 scissor, and free −OH, respectively, at the end of the polymer; Fig. 1B).
(
Synthesis and characterization of nanoparticles
The successful synthesis of blank and drug-loaded MPEG-PLA NPs was carried out by nano-precipitation along with solvent evaporation technique. TEM and FESEM images showed that synthesized NPs were spherical and smooth, with a mean diameter of around 70–220 nm (Fig. 2). The surface area of the particles analyzed by BET surface area analysis was estimated to be 14.491 sq.m/g, with a total pore volume of 0.0632 mL/g. This indicates a large surface area of the NP, along with sufficient pore volume to encapsulate an adequate quantity of drug.

(
Encapsulation of anti-leishmanial drugs
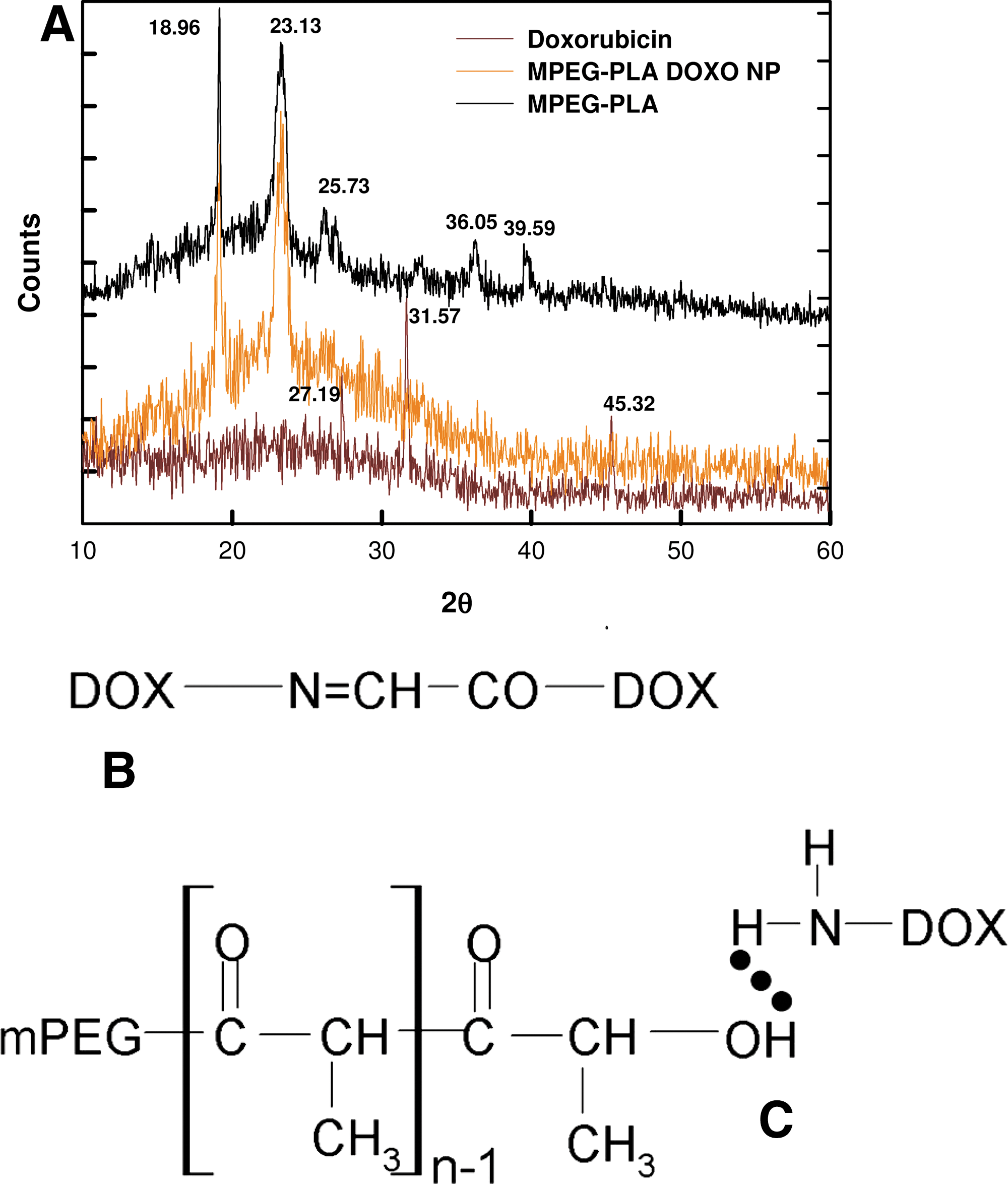
The data obtained through XRD were analyzed to confirm encapsulation of drug inside the MPEG-PLA NPs. Figure 3A shows XRD spectra of free drug (doxorubicin), encapsulated drug, and blank MPEG PLA NP. Peaks were obtained at 2θ of 27.19, 31.57, and 45.32 for crystalline doxorubicin, whereas these peaks disappeared in case of drug-loaded MPEG-PLA NPs, which confirmed the successful encapsulation of drug. In case of the blank NPs, some additional peaks at 25.73, 36.05, and 39.59 were obtained due to changes in crystal properties upon encapsulation of drug.
(
Enhanced uptake and slow release of encapsulated drug inside macrophages
Doxorubicin was found to yield absorption maxima at 485 nm, so the estimation of drug content was done at the same wavelength. The in vitro release profile of doxorubicin shows a moderately slow release of drug, with 84.59±7.32% release seen after 240 h (Fig. 4).
(
The in vitro release profile was also supported by intracellular release of doxorubicin as estimated by fluorescence spectroscopy. Epi-fluorescence images of macrophages showed bright red fluorescence of doxorubicin after just 8 h of incubation, with maximum intensity seen at 48 h for free doxorubicin, whereas for the encapsulated drug, only ? 5% macrophages exhibited fluorescence after 24 h, and the maximum intensity of doxorubicin fluorescence was delayed to 96 h (Fig. 5A–H). The same pattern was also obtained with fluorescence intensity parameters obtained by fluorescence spectrophotometry (Fig. 5I). Thus encapsulated doxorubicin is efficiently delivered to macrophages via NPs, and the release of the drug was found to be slow compared to native drug. These results indicate that encapsulated drug is released slowly inside the targeted cell, resulting in long-term exposure to the parasite, leading to enhanced efficacy.
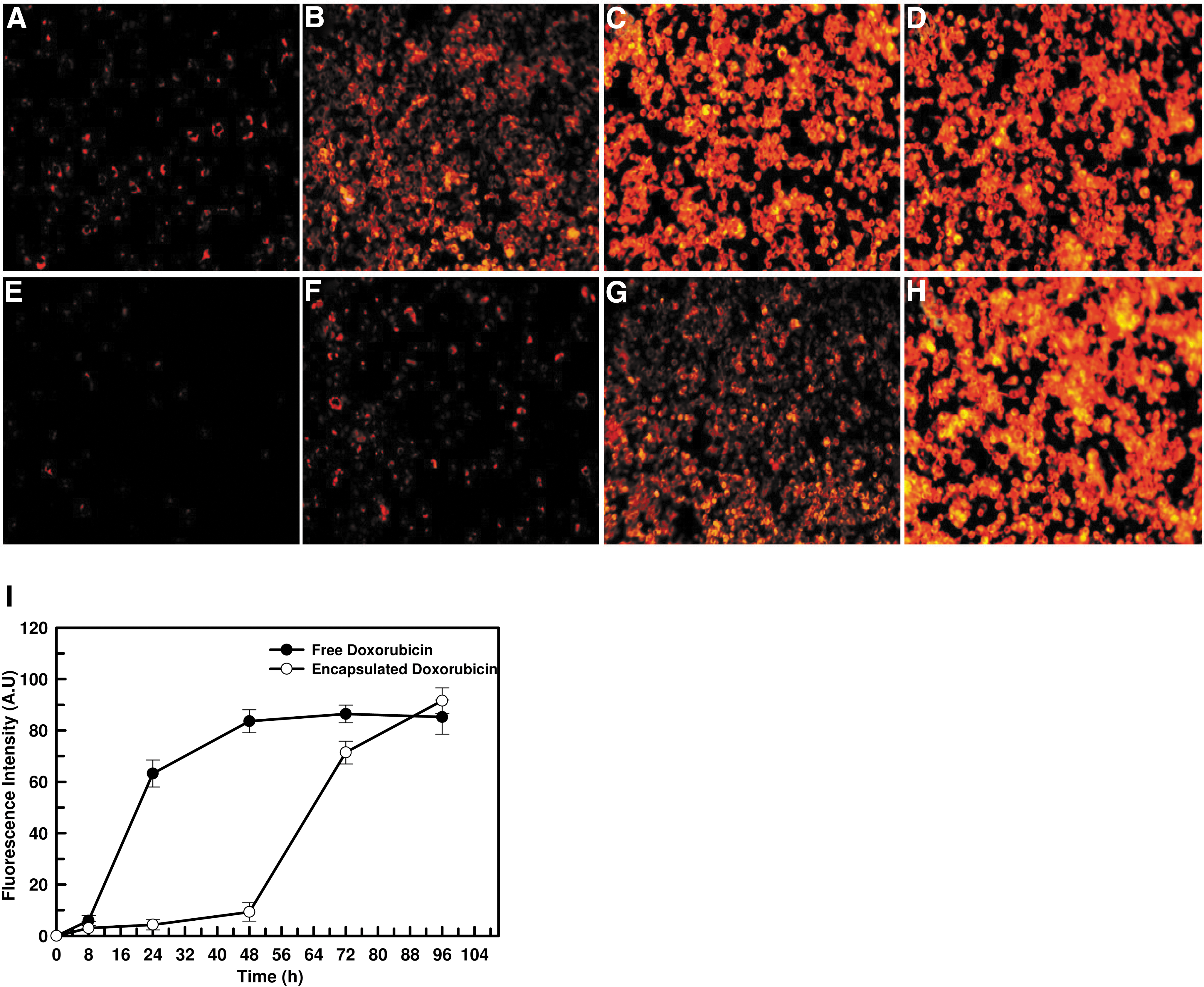
Fluorescence microscope images of macrophages at different time intervals of incubation with free doxorubicin (
Cellular toxicity and safety evaluation
Doxorubicin has been shown to have minor cardiotoxicity, and initially, mitomycin C was suggested to have pulmonary side effects. Mitomycin C does not increase the generation of oxygen radicals and cytokines (IL-6, TNF-α, or TGF-β) in pre-stimulated granulocytes in culture medium (Dirix et al. 1997). Macrophages are the parasite's proliferation site, and therefore are the main target for drug delivery. NP-mediated delivery is specific for macrophages, and thus it is important to evaluate the cytotoxicity of these formulations to the macrophages themselves. Cytotoxicity experiments were conducted on mouse macrophages (J774.1A). The results showed that drug toxicity was reduced after encapsulation. The percent viability of mouse macrophage (J774.1A) cell lines at 1.56 μM of free doxorubicin was 65.45±9.88%, whereas at the same concentration of encapsulated drug it was 78.87±6.99% (Fig. 6). In the case of mitomycin C, these values were found to be 83.82±16.14% and 92.78±4.92%, respectively. Thus one can conclude that polymeric encapsulation of doxorubicin and mitomycin C decreases their toxicity with increased therapeutic efficacy.
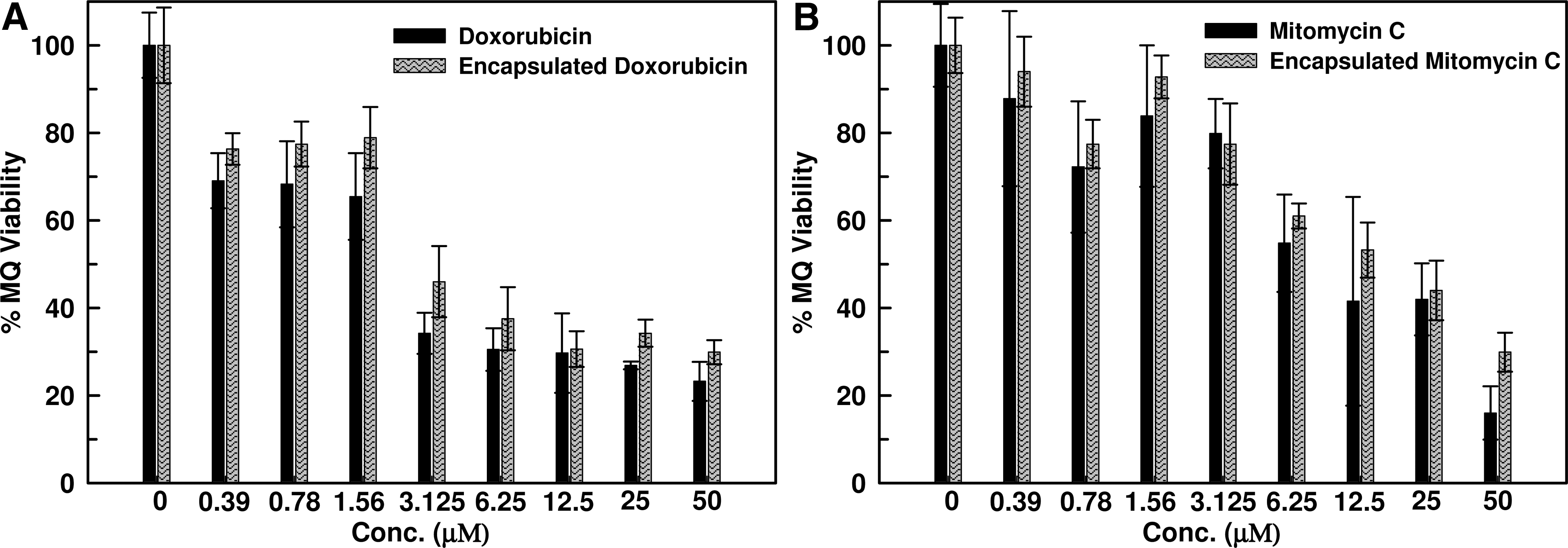
Cytotoxicity assay of free and encapsulated doxorubicin (
Discussion
Numerous techniques (liposomal formulations, micelles, emulsions, and NPs) have been utilized over the last decade to administer drugs to targeted tissues (Gref et al. 1995). Unfortunately, most of these modes of administration failed because of identification by polymorphonuclear cells (PMNs) and macrophages, and they were quickly phagocytized. However, this mechanism facilitates anti-leishmanial drug delivery to the target site (i.e., the macrophage itself). The biodegradable polymer MPEG-PLA seems to be appropriate for anti-leishmanial drug delivery due to its amphiphilic and biodegradable behavior, and its increased half-life in the bloodstream. Therefore, this idea prompted us to encapsulate doxorubicin and mitomycin C inside MPEG-PLA polymeric NP and evaluate its delivery to macrophages.
Doxorubicin is known to form doxo-doxo dimer in solution by a chemical reaction between 3′-NH2 and C9-α ketol side chain and Π-Π stacking of their planer ring (Menozzi et al. 1984; McLennan et al. 1985; Yokoyama et al. 1998). The dimeric form of doxorubicin is hydrophobic in nature, which enhances entrapment of doxorubicin in the PLA fraction (Fig. 3B). Alternatively, doxorubicin and mitomycin C are overall cationic molecules (due to −NH2 groups), whereas PLA is an ester having a terminal −OH group. This contributes to hydrogen bonding between drugs and PLA (Fig. 3C), thus enhancing entrapment of drugs in the core of the micelle. An important criterion to increase entrapment of these drugs in the micelle is to increase interaction between the drug and PLA core, which can be accomplished by increasing the amount of both drug and the PLA. Therefore we have used a MPEG-PLA polymer with a smaller fraction of PEG and more PLA.
Doxorubicin is protonated at its 3′-NH2 position and exists as 3′-NH3 +, which can interact with electronegative groups (Yi et al. 2005). According to a previous study (Righetti 1979), protonated behavior of the glycosidic amine is retained up to pH 7, and it is deprotonated above this point. Once the polymer enters the macrophages, 3′-NH2 of the drugs becomes protonated due to the acidic pH of the lysosomal compartment. This should facilitate the release of drug due to conversion of dimeric doxo-doxo to doxo monomer.
Conclusion
When drugs are administered in the body, only a small fraction reaches the macrophages, where the amastigote form of the parasite resides and multiplies. Thus for better efficacy of the drug, higher doses are required, which may result in toxic side effects. Preferential delivery of drug to macrophages is desirable after systemic or localized administration. Our study reports on the potential application of biodegradable polymer MPEG-PLA NPs loaded with doxorubicin and mitomycin C as a method for targeted drug delivery to macrophages. MPEG-PLA NPs loaded with doxorubicin and mitomycin C demonstrate targeted slow delivery to macrophages with few side effects.
Footnotes
Acknowledgments
We wish to acknowledge the infrastructural facilities provided by the Indian Institute of Technology Guwahati, and the financial support from the Department of Biotechnology (project no. BT/PR15211/MED/29/282/2011 and BT/01/IYBA/2009) of the Government of India in the form of a research grant to V.K.D.
Author Disclosure Statement
No conflicting financial interests exist.