
Abstract
Although Crimean-Congo hemorrhagic fever (CCHF) is a widespread tick-borne disease, little is known about its pathogenesis. The interaction of the virus with host cells is most likely responsible for the pathogenesis of CCHF. The main contributors are endothelial cells (ECs) and immune cells. There are 2 theories underlying the CCHF pathogenesis: One is that the virus interacts with the ECs directly and the other that it interacts indirectly via immune cells with subsequent release of soluble mediators. ECs are activated upon infection by the upregulation of soluble molecules and proinflammatory cytokines. Probably, in severe cases, deregulation and excessive release of the cytokines accompanied by endothelial activation have toxic effects, leading to increased vascular permeability, vasodilatation, and subsequently hypotension, multiple organ failure, shock, and death. Studies indicate that CCHF virus (CCHFV) also can impair the innate immune system and cause a delay in adaptive immune response, which is critical for the clearance of CCHFV. The virus has many different ways to block the immune response, leading to uncontrolled viral replication followed by systemic spread of the virus throughout the body. Partial activation of dendritic cells and macrophages, delayed induction of interferons, weak antibody response, apoptosis of lymphocytes, and hemophagocytosis are some of these tactics. However, there are many points waiting for clarification about the pathogenesis of CCHF. Although the high risk of contagiousness limits research, we need more studies to understand the CCHF pathogenesis better. Here we review the main characteristics of the pathogenesis of CCHF.
Introduction
CCHFV is a geographically widespread tick-borne virus ranging from southern areas of Africa and Eastern Europe to Russia and the Middle East (Vorou et al. 2007, Leblebicioglu 2010). The first documented outbreaks were recorded in 1944–1945 in Crimea during the Second World War and then the number of cases increased substantially with time (Whitehouse 2004). Humans are the only known host that develops disease after exposure to the virus. CCHFV also infects animals like cattle, sheep, goats, camels, and hares, but they remain asymptomatic. The major risk groups for acquiring CCHF are people living in endemic areas. They are exposed to infection from ticks of the genus Hyalomma, especially Hyalomma marginatum marginatum, or after handling of infected animals. Farmers, abattoir workers, veterinarians, and health care workers are included in the occupational risk groups (Whitehouse 2004, Vorou et al. 2007, Leblebicioglu 2010).
The incubation period of the disease ranges from a few days to 1 week, depending on the route of transmission and amount of the inoculum. The length is shorter for infections by tick bite or livestock contact compared to nosocomial infections (Vorou et al. 2007). The route of viral entry may also affect the severity of the disease. Mortality rates of nosocomially transmitted infections are much higher than the infections acquired by tick bites. This may be associated with high level of viremia in nosocomial infections (Altaf et al. 1998, Athar et al. 2005, Whitehouse 2004, Chinikar et al. 2010).
Initial symptoms are nonspecific and characterized by a rapid onset of fever, weakness, myalgia, headache, nausea, and vomiting lasting 3 days on average. The clinical spectrum varies from asymptomatic or mild infections to severe disease and death (Bodur et al. 2012). In severe cases, hemorrhagic manifestations develop 3–6 days after onset of the illness. Other signs include enlarged spleen and liver in approximately 30% of the patients. Thrombocytopenia, leukopenia, elevated liver enzymes, and prolonged bleeding times are the main laboratory features of CCHF (Vorou et al. 2007, Cevik et al. 2008, Hatipoglu et al. 2010). Convalescence starts on average at 15–20 days after onset of the disease and is characterized by weakness, lethargy, loss of hair, dizziness, labile pulse, hearing loss, and amnesia. All of these signs and symptoms are reversible, but may persist for more than a year (Whitehouse 2004).
Currently there is no specific antiviral therapy approved for CCHF (Whitehouse 2004). A recent meta-analysis revealed that ribavirin did not improve survival in CCHF (Ascioglu et al. 2011). Treatment options are limited, and supportive therapy is the best approach for managing the patients (Ozkurt et al. 2006, Elaldi et al. 2009, Bodur et al. 2011, Leblebicioglu et al. 2012). However, early administration of CCHFV hyperimmunoglobulin seems to be a promising new treatment approach (Kubar et al. 2011). The fatality rate of CCHF is very broad and ranges between 5% and 30%, depending on geographic regions and routes of entry (Jamil et al. 2005, Yilmaz et al. 2009). Death can occur due to multiple organ failure caused by severe anemia, dehydration, and shock (Swanepoel et al. 1989). The outcome of the disease has been shown to be significantly correlated with the amount of the virus in the blood, decreased platelet counts, elevated liver enzymes, prolonged bleeding times, decreased fibrinogen levels, somnolence, and gastrointestinal bleeding (Swanepoel et al. 1989, Ergönül et al. 2006a, Cevik et al. 2007, Duh et al. 2007, Cevik et al. 2008, Hatipoglu et al. 2010) (Fig. 1). Additionally in the study of Akinci et al., a relationship between HLA alleles and severity of the disease was detected. The frequency of HLA-A*23 was found significantly higher in severe cases (unpublished data). However, there is no obvious correlation between genetic diversity of the virus and the pathogenicity for humans (Deyde et al. 2006). Due to a lack of an effective vaccine for CCHF, avoiding exposure to CCHFV is the most efficient way for the prevention and control of the disease (Whitehouse 2004, Vorou et al. 2007).
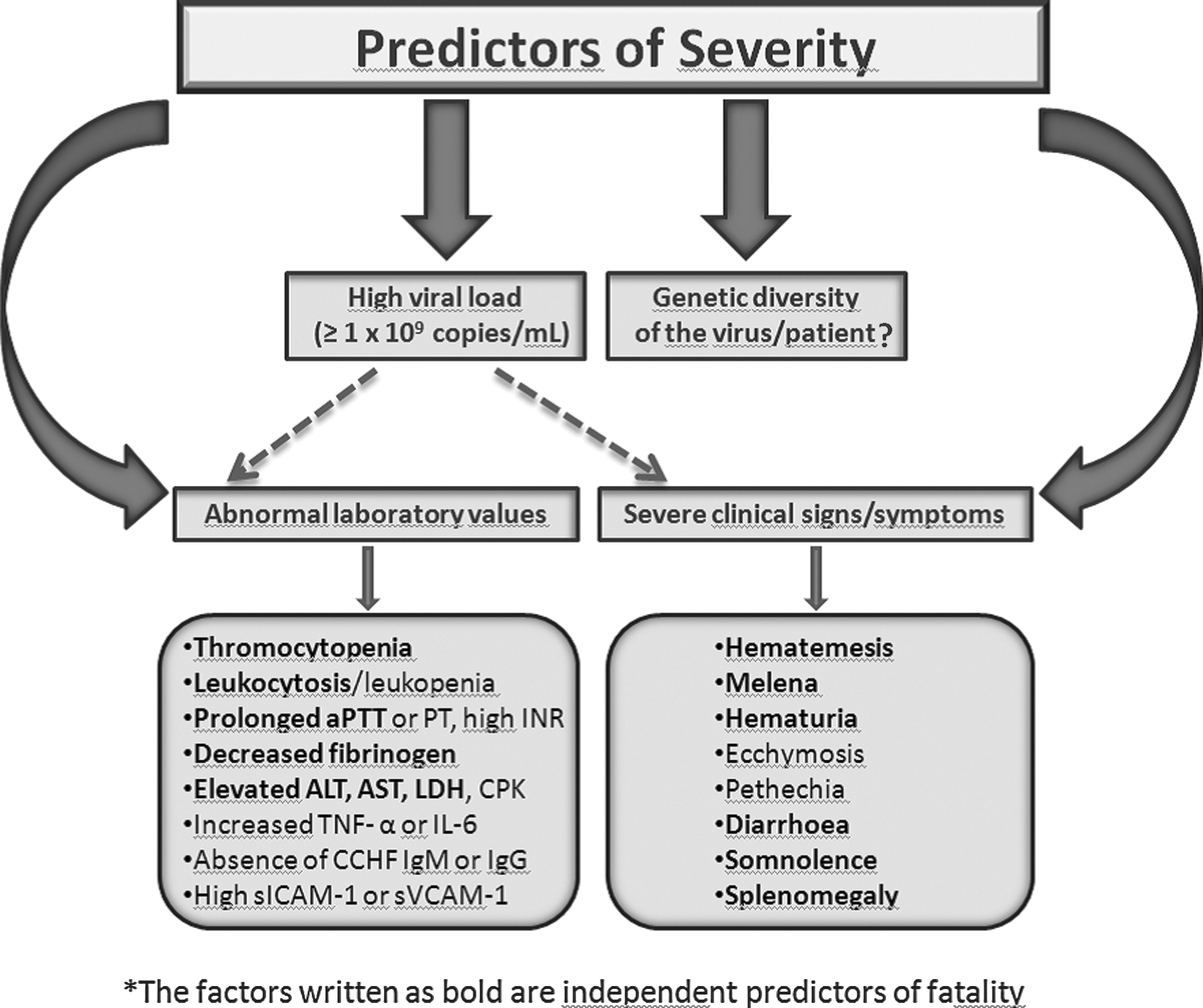
Severity criteria of CCHF infection. aPTT, activated partial thromboplastin time; PT, prothrombin time; ALT, alanine aminotransferase; AST, aspartate aminotransferase; LDH, lactate dehydrogenase; CPK, creatine phosphokinase; TNF-α, tumor necrosis factor-α; IL-6, interleukin-6; CCHG, Crimean-Congo hemorrhagic fever; Ig, immunoglobulin; ICAM-1, intercellular adhesion molecule-1; VCAM-1, vascular cell adhesion molecule-1.
Pathogenesis
Although CCHFV is a widely distributed tick-borne virus, little is known about the pathogenesis of CCHF. Due to the necessity of biosafety level 4 (BSL-4) laboratories for handling the virus, lack of animal models and the occurrence of infections in areas where facilities are limited to conduct research, CCHF pathogenesis is poorly understood. The limited knowledge originates mostly from blood analyses, autopsies, and liver biopsies of patients (Whitehouse 2004). The interaction between the virus and host cells is most likely responsible for the pathogenesis of CCHF. The main contributors are endothelial cells (ECs) and immune cells (Fig. 2).
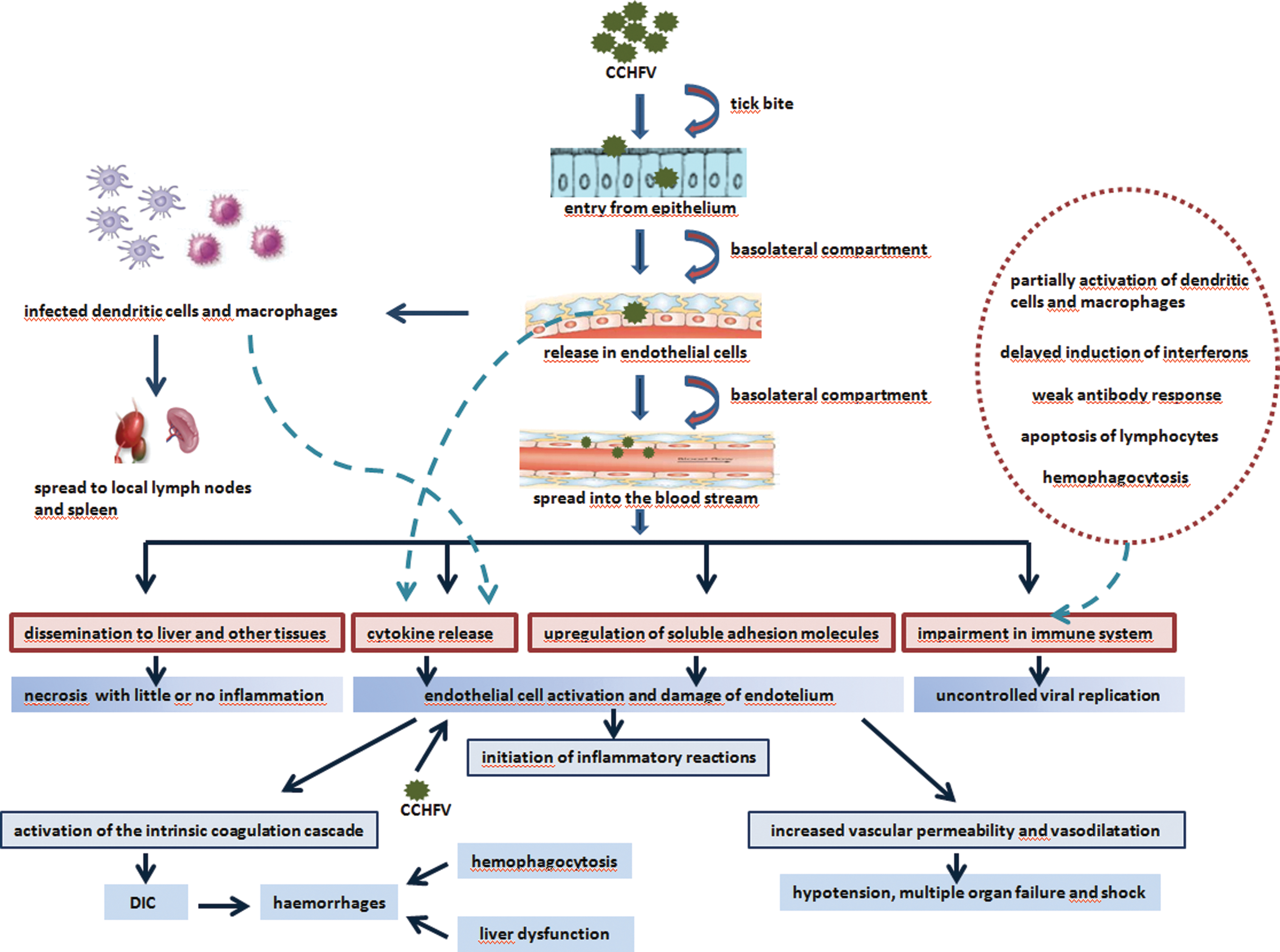
Pathogenesis of CCHF. Endothelium is the major target of CCHFV and can be activated directly by the virus and/or indirectly by virus-induced soluble mediators. Activation of endothelial cells is critical to start the inflammatory reactions, increase of vascular permeability, and activation of the intrinsic coagulation cascade. The virus also has many different ways to block the immune response permitting uncontrolled viral replication. CCHF, Crimean-Congo hemorrhagic fever; CCHFV, CCHF virus; DIC, disseminated intravascular coagulation.
Viral entry and release in ECs
The first barrier is the epithelium, and the virus probably overcomes it with the help of the tick bite. Viral attachment proteins are localized in the basolateral membrane. Viral entry and release in ECs most likely occur via the basolateral compartment. As the basolateral compartment of ECs is directed toward the blood vessels, viral release from the basolateral membrane into the bloodstream causes systemic dissemination (Connolly-Andersen 2010). Entry of the virus into host cells is mediated by the binding of the envelope glycoprotein Gc to cell-surface-associated receptors. Human cell-surface nucleolin is suspected to be a CCHFV entry factor (Xiao et al. 2011).
Dissemination of the virus
The dissemination route of the CCHFV to the body is not exactly known. The tick bite probably causes viral release into the vascular system. Amplification of the virus in tissue resident macrophages and dendritic cells (DCs) may facilitate transmission of the virus to local lymph nodes, spleen, and finally to systemic circulation of the host (Connolly-Andersen 2010).
In animal models, it has been demonstrated that CCHFV replicates in blood, liver, and spleen at the beginning, and then spreads systemically to lung, kidney, and brain. Early viral replication occurs in the blood on the first day of infection and subsequently in the liver and spleen on the second day (Bente et al. 2010). The liver is an important target for CCHFV. It is easy for the virus to get to the basolateral plasma membranes of hepatocytes and ECs because of the fenestrated liver sinusoids and the lack of a basement membrane (Connolly-Andersen 2010). Involvement of the brain is at a later stage of the infection (Bente et al. 2010). Transmission of the virus to the cerebrospinal fluid (CSF) was demonstrated in other viral hemorrhagic fevers as a result of increased vascular permeability by cytokine release and subsequently disruption of the blood–CSF barrier (Chaturvedi et al. 1991, Gunther et al. 2001, Domingues et al. 2008, Kang et al. 2010).
Endothelial damage
The main clinical characteristics of CCHF, such as hemorrhage and increased vascular permeability and presence of viral antigens in ECs, indicate that the endothelium is the major target of CCHFV (Burt et al. 1997, Bodur et al. 2010, Ozturk et al. 2010). Activation of ECs is critical for starting the inflammatory reactions involving leukocyte rolling, adhesion, and transmigration into inflamed areas as well as organization of the immune response to infection and increase of vascular permeability (Connolly-Andersen 2010, Connolly-Andersen et al. 2011). Endothelial damage can also contribute to activation of the intrinsic coagulation cascade (Weber and Mirazimi 2008). According to the classical theories, the endothelium can be activated directly by the virus and/or indirectly by virus-induced host-derived soluble mediators (Schnittler and Feldmann 2003, Connolly-Andersen 2010, Connolly-Andersen et al. 2011).
Upregulation of soluble adhesion molecules
ECs are activated upon infection by the upregulation of soluble molecules involving E-selectin, vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), and an increased adhesion of leukocytes in response to inflammatory mediators. However, it is unclear if those molecules are derived from ECs or from leukocytes (Connolly-Andersen 2010, Connolly-Andersen et al. 2011). Soluble adhesion molecules ICAM-1 and VCAM-1 can be used as markers of endothelial activation and as indicators of vascular damage and disease severity (Leeuwenberg et al. 1992, Videm et al. 2008, Bodur et al. 2010, Ozturk et al. 2010).
Cytokine release
Main contributors in CCHF progression are the proinflammatory cytokines interleukin (IL)-1, IL-6, IL-8, IL-10 and tumor necrosis factor-α (TNF-α). While resting ECs showed weak constitutive production of cytokines, an increased release of these molecules was observed upon infection (Connolly-Andersen et al. 2011). Besides ECs, these proinflammatory cytokines are also excreted from CCHFV-infected macrophages and DCs (Connolly-Andersen 2010). It was determined that soluble mediators released from CCHFV-infected DCs had the ability to enhance ICAM-1 expression (Connolly-Andersen et al. 2009, Connolly-Andersen 2010, Connolly-Andersen et al. 2011). Interestingly, addition of anti-TNF-α antibody prevented the ICAM-1 upregulation effectively and indicated that TNF-α may be the major cytokine leading to activation of ECs (Connolly-Andersen 2010).
In several studies, high serum levels of the proinflammatory cytokines IL-6, IL-8, IL-10, and TNF-α were implicated as prognostic factors showing disease severity in CCHF patients (Ergonul et al. 2006b, Papa et al. 2006, Connolly-Andersen et al. 2009, Bente et al. 2010, Saksida et al. 2010). Probably, in severe cases, deregulation and excessive release of the cytokines have toxic effects on the endothelium leading to increased vascular permeability, vasodilatation, multiple organ failure, and shock (Connolly-Andersen 2010). Thus, the pathogenesis of CCHF and sepsis is similar. In both of them, deregulated cytokine storm leads to a systemic vascular collapse.
Effect of viral dose
A positive correlation between a high viral load and the severity of the CCHF infection was demonstrated (Cevik et al. 2007, Duh et al. 2007, Saksida et al. 2010). Viral loads ≥1×109 RNA copies/mL were strongly associated with fatality in CCHF patients (Cevik et al. 2007). While the viral titres were increasing with time in fatal cases, a decrease was observed in patients who survived (Saksida et al. 2010). Activation of ECs and leukocyte adhesion were found to be dependent on viral dose circulating in the bloodstream. Increasing CCHFV titers resulted in enhanced ICAM-1 levels (Connolly-Andersen 2010, Saksida et al. 2010, Connolly-Andersen et al. 2011). A positive correlation between the viral load and the cytokine levels was also observed (Saksida et al. 2010). Thus, the mechanism underlying the association between the viral load and the severity of disease may be due to high cytokine levels and enhanced endothelial activation at the high viral titers.
Increased vascular permeability
The exact mechanism of increased vascular permeability in CCHF is unknown. The release of vasoactive mediators by activated ECs is likely to be responsible for increased vascular permeability (Connolly-Andersen 2010, Connolly-Andersen et al. 2011). TNF-α possibly causes the vascular leakage in CCHF. When ECs are exposed to TNF-α, vascular permeability increases by a mechanism involving the destabilization of microtubules (Petrache et al. 2003, Connolly-Andersen et al. 2007).
Role of the tight junctions
Tight junctions (TJs) separate the plasma membrane of epithelial cells and regulate cellular permeability. Disruption of junctional complexes is followed by increased permeability and leakage. It was shown that CCHFV replication has no direct effect on epithelial permeability and on the localization of the TJ proteins occludin and ZO-1 in epithelial cells (Connolly-Andersen 2010). Thus, increased vascular permeability in CCHF does not seem to be caused by direct viral interaction with TJ proteins.
Levels of vascular endothelial cadherin
Vascular endothelial cadherin (VE-cadherin) is an important component of adherens junctions (Venkiteswaran et al. 2002, Vincent et al. 2004). In a study analyzing the localization of VE-cadherin, no significant changes in the levels of VE-cadherin mRNA or protein were observed between noninfected and CCHFV-infected ECs (Connolly-Andersen et al. 2011).
Impairment in the immune response
The innate immune response is the first step of defense against viruses. Studies indicate that CCHFV can impair the innate immune system and cause a delay in the adaptive immune response, which is critical for clearance of CCHFV (Weber and Mirazimi 2008, Bente et al. 2010, Peyrefitte et al. 2010, Saksida et al. 2010). The virus has many different ways to block the immune response leading uncontrolled viral replication and the systemic spread of the virus throughout the body.
Partial activation of macrophages and DCs
Macrophages and DCs play an important role at very early stages of the CCHF infection (Peyrefitte et al. 2010). They have an influential role in the initiation and control of the adaptive immune system by releasing proinflammatory cytokines and chemokines (Connolly-Andersen et al. 2009, Peyrefitte et al. 2010). However, in vitro and in vivo studies demonstrated that these cell types partially matured to gain the capability to function as an antigen-presenting cell, and this resulted in a lack of upregulation of major histocompatibility complex II (MHC II), which is an important protein for the priming of naïve T cells. (Bente et al. 2010, Peyrefitte et al. 2010).
Hemophagocytosis
Hemophagocytic syndrome is a rare and severe disease associated with the hyperactivation of monocytes and macrophages, causing cytopenias as a result of an excessive phagocytosis of blood cells (Favara 1992, Fisman 2000, Tasdelen Fisgin et al. 2008). It is characterized by fever, hepatosplenomegaly, cytopenia, and hemophagocytosis in bone marrow, liver, and lymph nodes (Karti et al. 2004, Cagatay et al. 2007). Hemophagocytic syndrome has been detected in up to 50% of the patients with CCHF and mostly demonstrated in the patients with severe bleeding (Karti et al. 2004, Cagatay et al. 2007, Tasdelen Fisgin et al. 2008).
Delayed induction of interferons
Interferons (IFNs) are important compounds for the antiviral response of the innate immune system and have important roles in limiting the spread of the infection. Production of type 1 IFNs (IFN-α and -β) yields a rapid and efficient host response against the viruses (Weber and Mirazimi 2008). Activation of the innate immune system by the molecules produced during viral replication leads to induction and secretion of type I IFNs and subsequently upregulation of interferon-stimulated genes (ISGs) (Samuel 2001, Andersson et al. 2008). The interferon regulatory factor-3 (IRF-3) plays a key role in the induction of IFNs. Therefore it is a common target for most of the viruses and many of them induce IRF-3 in the early stage of infection (Andersson et al. 2008). IFNs have antiviral effects by inducing expression of potent antiviral proteins. Moreover, they have other roles such as inhibition of cell proliferation, immunomodulation, and regulation of apoptosis (Weber and Mirazimi 2008, Connolly-Andersen 2010).
CCHFV is one of the IFN-sensitive viruses (Andersson et al. 2008, Weber and Mirazimi 2008, Bereczky et al. 2010). However, it delays production of IFNs in infected cells and prevents the antiviral effect of these molecules. In a recent study, CCHFV titers decreased significantly when the cells were treated with IFN-α 24 h before the infection. On the other hand, when IFN-α was administered to infected cells 6 h after the infection, no effect on the viral titers was detected. This study indicated that IFNs are inefficient in controlling the viral replication in established CCHF infection (Andersson et al. 2008). Interferon-induced MxA protein expression also plays an important role in the antiviral activity of IFNs against CCHFV via interaction with CCHFV nucleocapsid protein and subsequently inhibition of virus replication (Weber and Mirazimi 2008, Andersson et al. 2008). However, it is suggested that the MxA protein may be induced insufficiently or too late to prevent the progression of the disease in CCHF-infected patients (Andersson et al. 2008).
CCHFV has mechanisms of protection from IFNs, like other viruses. It has been suggested that CCHFV delays induction of IFNs by processing the viral 5′ RNA termini, hence avoiding RNA helicase RIG-I stimulation and IFN transcription (Connolly-Andersen 2010). It was also determined that replicating CCHFV stimulates interferon regulatory factor-3 (IRF-3) nuclear translocation late during the infection and causes a delay in the IFN response by intervening in the activation pathway of IRF-3. Thus, it allows the virus to replicate in the first few hours of CCHF infection (Andersson et al. 2008). Additionally, CCHFV strongly counteracts IFN signaling and the induction of IFN synthesis in several ways. One of them is that CCHFV does not produce significant amounts of the IFN inducer molecule double-stranded RNA (dsRNA) similar to other negative-strand RNA viruses (Weber et al. 2006, Weber and Mirazimi 2008). The other is that the RIG-I signaling pathway, which is triggered by a viral single-strained RNA (ssRNA) carrying a 50 triphosphate, is not activated by the CCHFV genome (Habjan et al. 2008, Weber and Mirazimi 2008).
In animal models, CCHF infection was evaluated in mice deficient in IFN signaling pathways. Although CCHF infection did not develop in immune-competent mice, rapid development of the disease leading to death was observed in mice with no IFN response. Thus, it was hypothesized that IFN has an antiviral effect against CCHF, but the ability of CCHFV to disable the IFN response is human specific (Bente et al. 2010). IFN induction by CCHFV-infected cells is weak and relatively late in humans (Weber and Mirazimi 2008, Andersson et al. 2008). However, in animals, with efficient IFN response, CCHF infection is weakened and the disease does not occur (Bente et al. 2010).
Undetectable antibody response
Antibody production against CCHFV is 1 of the important factors for survival. Fatal CCHF cases rarely established an antibody response, and a significant reverse correlation between viral load and antibody levels was observed (Duh et al. 2007, Wölfel et al. 2007, Saksida et al. 2010). The association between a weaker or undetectable antibody response and a higher viral load in fatal cases indicates that an impaired immune response leads to uncontrolled replication of the virus. Although it is not exactly known, the underlying mechanism could be as a result of inadequate activation of the adaptive immune response by DCs because of partial maturation of CCHF-infected DCs (Bente et al. 2010, Peyrefitte et al. 2010).
Depletion in the numbers of natural killer cells and lymphocytes
Natural killer (NK) cells are part of the innate immune system response against viruses and play a role in the detection and lysis of infected cells (Yilmaz et al. 2008). T and B lymphocytes also have important functions in the immune response. In a recently developed mouse model for CCHF, it was shown that the lymphocytes in the spleen were activated within 3 days. The numbers of NK, B, and T cells increased on the first day of infection, but they decreased in numbers significantly on the third day, regardless of activation (Bente et al. 2010). Possibly, these cells increased because of the early production of cytokines, but later the uncontrolled apoptosis of lymphocytes contributes to a depletion in lymphocyte counts both in the blood and the spleen, which is presented as lymphopenia (Geisbert et al. 2000, Bente et al. 2010, Connolly-Andersen 2010). Therefore, both innate and adaptive immune systems are disrupted, leading to an inability to control early viral replication.
The association with peripheral lymphocytes and the severity of the disease has been investigated in several studies, but conflicting results have been obtained. One study found a positive correlation between disease severity and total counts of NK cells. However, no significant difference was found in the numbers of T and B lymphocytes between severe and nonsevere cases. (Yilmaz et al. 2008). On the other hand, another study presented no significant difference in NK cells between fatal and survived cases, whereas cytotoxic T cells were significantly higher in fatal cases and a correlation was detected with higher viral load (Akinci et al. 2009).
There are also ongoing studies about the effect of CCHFV on the immune system. In a recent study performed by Bastürk et al., increased expression of human leukocyte antigen-G (HLA-G) molecules on the surface of immune cells was determined during CCHFV infection, leading to depression in the immune response of the host (unpublished data).
Coagulation Disorders and Bleeding
Hemorrhage is one of the main signs of CCHF. Hemorrhagic fever viruses modify hemostasis using 2 mechanisms. The first mechanism is a direct effect on cells involved in hemostasis, like thrombocytes and ECs. The second mechanism is through immunological and inflammatory pathways leading to cell injury (Chen and Cosgriff 2000). In CCHF, vascular endothelial injury, disseminated intravascular coagulation (DIC), thrombocytopenia, liver dysfunction, and diminished levels of coagulation factors lead to hemorrhage (Burt et al. 1997, Chen and Cosgriff 2000, Peters and Zaki 2002).
Endothelial damage stimulates platelet aggregation and degranulation, followed by activation of the intrinsic coagulation cascade and consequently DIC (Schnittler and Feldmann 2003). DIC is an important cause of hemorrhages and results in an increased consumption of both coagulation factors and platelets (van Gorp et al. 1999). It has been reported in severe CCHF cases and is associated with a poor prognosis (Whitehouse 2004, Ergonul et al. 2006b).
Thrombocytopenia is a consistent feature of CCHF, and a platelet count of 20×103/L or below is an independent predictor of fatality (Cevik et al. 2008). Decreased platelet production due to bone marrow hypoplasia or increased platelet consumption caused by DIC leads to thrombocytopenia (Chen and Cosgriff 2000).
Diminished levels of plasma coagulation factors result from either increased consumption in DIC or impaired synthesis as a consequence of liver dysfunction (Chen and Cosgriff 2000). It is presented in clinical practice as prolonged prothrombin time (PT) and activated partial thromboplastin time (aPTT) values and high international normalized ratio (INR) levels. Severity of the disease is strongly correlated with the degree of hemostatic disorders (Swanepoel et al. 1989, Bakir et al. 2005, Ergonul et al. 2006a, Cevik et al. 2008). In a recent study, plasma levels of thrombin activatable fibrinolysis inhibitor (TAFI) activity were found to be significantly lower in CCHF patients compared to healthy controls (Sonmez et al. 2007). TAFI is a proenzyme synthesized in liver and stabilizes the fibrin clot by downregulation of fibrinolysis (Bouma and Meijers 2004, Sonmez et al. 2007).
Histopathologic Findings
Histopathologic studies are limited in CCHF cases. Marked histopathologic changes occurred in the liver and spleen, which are the main sites of CCHFV replication. CCHFV exhibits hepatotropism and rapidly replicates to high titers in hepatocytes, which illustrates the important role of the liver for the amplification of the virus (Bente et al. 2010, Rodrigues et al. 2012). During replication, the virus also induces a cytopathic effect, which may be an in vitro marker of CCHFV pathogenicity (Rodrigues et al. 2012). Immunohistochemical studies in autopsies and animal models showed infection of Kupffer cells, hepatic endothelial cells, and hepatocytes. A variable degree of hepatocellular necrosis, Kupffer cell hyperplasia, fatty change, and mononuclear portal inflammation was found in an examination by microscopy (Burt et al. 1997). Generally, necrosis of hepatocytes existed in multiple foci, ranging from mild to severe necrosis with little or no inflammatory infiltrates (Baskerville et al. 1981, Burt et al. 1997, Bente et al. 2010). The necrotic areas were marked by hemorrhage and associated with the eosinophilic change of hepatocytes and Councilman bodies (Burt et al. 1997). In addition, the extent of necrosis correlated with an elevation of liver enzymes (alanine aminotransferase [ALT], aspartate aminotransferase [AST]), which are prognostic factors of fatality (Swanepoel et al. 1989, Burt et al. 1997, Bakir et al. 2005, Ergonul et al. 2006a, Cevik et al. 2008, Hatipoglu et al. 2010). Steatosis, fatty degeneration, hepatocyte swelling, intercellular edema, and hemorrhage were also demonstrated in other viral hemorrhagic fevers (Paes et al. 2005, Paes et al. 2009, Warfield et al. 2009).
CCHFV induces apoptosis of hepatocytes by altering both intrinsic and extrinsic pathways. A comparison between CCHFV and a nonpathogenic Nairovirus for humans demonstrated that the most prominent difference was the absence of cytopathic effect and apoptosis in liver cells (Rodrigues et al. 2012). CCHFV-induced apoptosis involves the mitochondrial pathway and is able to cause endoplasmic reticulum (ER) stress. Viral replication-associated products may be critical to elicit the apoptosis. Decreased expression of Bcl-2 proteins probably initiates the intrinsic apoptotic pathway (Rodrigues et al. 2012).
The spleen is another important target for the virus. The pathological findings of spleen involve prominent splenic lymphoid apoptosis and depletion accompanied by karyorrhectic debris, splenocyte necrosis, and dilated sinusoids (Baskerville et al. 1981, Bente et al. 2010). The endothelial cells were also infected. The infection consisted of hemorrhages, edema, and/or necrosis without inflammatory infiltrates (Baskerville et al. 1981, Joubert et al. 1985, Burt et al. 1997, Connolly-Andersen 2010).
Viral antigens could be detected in spleen and liver cells (Bente et al. 2010). Macroscopic examination revealed a discolored liver and spleen with serosal petechia (Bente et al. 2010). The other findings in other tissues included interstitial pneumonia and intestinal hemorrhages. Diffuse alveolar damage and intraalveolar hemorrhage were present in the lungs (Burt et al. 1997).
Conclusion
CCHF is an important emerging infectious disease with limited treatment alternatives. Highlighting the pathogenesis of the disease may lead to new investigations for developing novel therapeutics. Endothelial damage and impaired immune response seem to play an important role in clinical progression and the severity of the disease. Excessive release of cytokines and soluble molecules activates endothelial cells, leading to increased vascular permeability, vasodilation, and subsequently hypotension, multiple organ failure, and shock. Delayed induction of IFNs, a weak antibody response, and the partial maturation of macrophages and DCs cause uncontrolled replication of the virus. Thus, administration of specific immunoglobulins may be effective in the treatment of CCHF. On the other hand, inhibition of cytokine release, as in sepsis or the early application of IFNs before the development of the infection, could be evaluated as an alternative treatment. However, there are many points waiting to be clarified concerning the pathogenesis of CCHF. Although the high risk of contagiousness limits the investigations, animal models may help to understand the CCHF pathogenesis better.
Author Disclosure Statement
No competing financial interests exist.