
Abstract
The Borrelia burgdorferi sensu lato (s.l.) group comprises genetically related spirochetes, mostly associated with tick species belonging to the Ixodes ricinus complex in the Northern Hemisphere. The present study evaluated borrelial infection in the tick Ixodes pararicinus, which is the only representative species of the I. ricinus complex in Uruguay. A total of 137 I. pararicinus ticks were collected from deer, cattle, or vegetation in 2 Uruguayan Departments. A part of these ticks was tested directly by PCR targeting the borrelial gene flagellin (fla), whereas another part of the ticks was inoculated into Barbour–Stoenner–Kelly (BSK)-H medium in an attempt to isolate Borrelia. Overall, Borrelia infection was detected in 9 males and 1 nymphal tick pool. These ticks were found to be infected by unique fla haplotypes, which were shown through phylogenetic analysis to represent possibly 2 new B. burgdorferi s.l. genospecies, 1 associated with B. bissettii, the other phylogenetically closest to B. americana. These results were reinforced by PCR and DNA sequencing analyses of portions of 2 additional borrelial genes, rrfA-rrlB intergenic spacer region (IGS) and 16S rDNA (rrs). Weekly examinations of BSK cultures by dark-field microscopy failed to demonstrate live Borrelia through a 100-day incubation period. However, Borrelia DNA was detected by fla-PCR in culture media from 2 vials up to 90 days after inoculation. To the best of our knowledge, this is the first report of B. burgdorferi s.l. infecting ticks in South America.
Introduction
The ecology, global distribution, and phylogeny of spirochetes comprising the B. burgdorferi s.l. group are inexorably related to the biology of their natural hosts and vectors, which in most of the cases are the ixodid ticks of the Ixodes ricinus complex (Korenberg et al. 2010), including 18 species (Venzal et al. 2005). In the Western Hemisphere, the I. ricinus complex is represented by 8 species, namely the Nearctic species I. jellisoni, I. muris, I. pacificus, and I. scapularis; the Neotropical I. aragaoi and I. pararicinus; and the Nearctic–Neotropical I. affinis, and I. minor (Venzal et al. 2005). Among these, I. scapularis and I. pacificus are primary vectors of Lyme disease agents in North America, whereas some of the other Nearctic species are involved in the enzootic cycles of B. burgdorferi s.l. (Dolan et al. 2000, Maggi et al. 2010, Scott et al. 2010). Regarding South America, the only study that searched for borrelial infection in ticks of the I. ricinus complex reported no infection among 2600 specimens of I. affinis and I. pararicinus ticks from Colombia (Mattar and Lopez Valencia 1998).
I. pararicinus is the only existing species of the I. ricinus complex in Uruguay, where adult ticks usually feed on wild deer and domestic cattle, and the tick immature stages feed on passerine birds and sigmodontinae rodents (Venzal et al. 2005). This host usage pattern is typical for the vectors of Lyme disease in the Northern Hemisphere (Stanek et al. 2012). However, human infestation by I. pararicinus has never been reported (Venzal et al. 2005, Guglielmone et al. 2006). Besides Uruguay, I. pararicinus is known to occur in Argentina, Colombia, and Peru (Blair et al. 2004, Venzal et al. 2005). However, recent studies suggest that different species might be represented under the taxon I. pararicinus in its current distribution area (J.M.V., unpublished data). In addition, the species I. aragaoi, known to occur only in Brazil, is possibly conspecific to the Uruguayan populations of I. pararicinus, a condition yet to be formally confirmed (Venzal et al. 2005).
Because the B. burgdorferi s.l. group is primarily associated with ticks of the I. ricinus complex (Korenberg et al. 2010), I. pararicinus ticks are indeed good candidates to harbor this bacterial group in South America. In the present study, we demonstrate that I. pararicinus ticks from Uruguay were infected by different genotypes of B. burgdorferi s.l. To the best of our knowledge, this is the first report of B. burgdorferi s.l. infecting ticks in South America and also the first demonstration of this important bacterial group in a tick species endemic to the southern hemisphere.
Materials and Methods
Ticks
Four tick collections (TC1–TC4) were performed from deer (Mazama gouazoubira), domestic cattle, or vegetation in Uruguay from 2009 to 2011, totaling 137 ticks (Table 1). TC1 ticks (all collected from a single deer) and TC2 ticks (collected from various cows) were immediately preserved in 70% ethanol and sent to the laboratory for molecular analyses (procedures described below). TC3 ticks (collected from cows) and TC4 ticks (collected from vegetation) were kept alive for 1–2 days before being inoculated into culture medium, as described below. All ticks were morphologically identified as I. pararicinus according to Keirans et al. (1985) and Venzal et al. (2005). In addition, taxonomic identification of 2 ticks was confirmed by molecular tools through DNA sequencing a portion of the tick mitochondrial 16S rRNA gene, as previously described (Ogrzewalska et al. 2009).
See Fig. 1 for phylogenetic placement of the fla haplotypes detected in the present study.
Ticks were tested in 1 pool of 11 males (tick collection 3) or 2 pools of 5 and 6 nymphs each (tick collection 4). In this case, 1 pool of males and 1 pool of nymphs were positive. For calculation purposes, we considered that only 1 tick specimen was infected in each positive pool.
Borrelia molecular identification was performed in Barbour–Stoener–Kelly (BSK) medium inoculated with tick pools.
Molecular analysis for Borrelia
Ethanol-preserved ticks were left at room temperature for a minimum of 30 min for ethanol evaporation and subjected individually to DNA extraction by the guanidine isothiocynate–phenol technique (Sangioni et al. 2005). They were tested for the presence of Borrelia DNA by nested-PCR targeting the flagellin gene (fla) of Borrelia spp. using primers FlaLL (5′-ACA TAT TCA GAT GCA GAC AGA GGT- 3′) and FLA RL (5′-GCA ATC ATA GCC ATT GCA GAT TGT-3′) for the first reaction, and primers FlaLS (5′-AAC AGC TGA AGA GCT TGG AAT G-3′) and FLA RS (5′-CTT TGA TCA CTT ATC ATT CTA ATA GC-3′) for the nested reaction. The first reaction targets a 665-bp fragment, and the nested reaction targets a 354-bp fragment. PCR conditions were adopted as described elsewhere (Barbour et al. 1996). Ticks positive by the fla gene PCR were further tested by 2 additional PCR protocols, 1 targeting a 225- to 255-bp fragment of the rrfA-rrlB intergenic spacer region (IGS) using primers IGSb (5′-GTT AAG CTC TTA TTC GCT GAT GGT A-3′) and IGSa (5′-CGA CCT TCT TCG CCT TAA AGC-3′), as previously described (Derdáková et al. 2003); and another protocol targeting a 746-bp of the 16S rDNA (rrs) using primers S5-F (5′-GAG GAA TAA GCT TTG TAG GA-3′) and S13-R (5′-GAC GTC ATC CTC ACC TTC CT-3′) (Le Fleche et al. 1997). For all PCR reactions, water was included as negative control, and Borrelia anserina DNA (Ataliba et al. 2007) was included as positive control. PCR products were purified using ExoSAP (USB Corporation, Cleveland, OH), and submitted to DNA sequencing in an automatic sequencer (Applied Biosystems/PerkinElmer, Foster City, CA) according to the manufacturer`s protocol. DNA sequences were aligned and compared to each other by using EditSeq and MegAlign sequence tools (DNASTAR, Madison, WI), and also compared to corresponding sequences in GenBank through BLAST analyses (
Attempts to isolate Borrelia
Ticks brought alive to the laboratory were surface-sterilized through immersion in iodine/70% ethanol for 10–15 min and rinsed with sterile phosphate-buffered saline (PBS). Individual (females) or pooled (5–6 males or nymphs) ticks were minced and placed directly into tubes containing 4 mL of Barbour–Stoenner–Kelly (BSK)-H medium, complete (Sigma-Aldrich, St. Louis, MO), supplemented with kanamycin (100 μg/mL) and amphotericin B (2.5 μg/mL). The cultures were incubated at 34°C for 100 days and examined weekly by dark-field microscopy for the presence of live spirochetes. In addition, 1-mL aliquots of culture medium were taken off from all tubes 15 days after inoculation, and divided into 0.5 mL for a blind passage into new tubes containing BSK-H medium without antibiotics, and 0.5 mL for DNA extraction by washing through centrifugation and boiling at 100°C for 10 min. New DNA extracts were generated from the original tick-inoculated tubes at 30, 45, and 90 days after inoculation, and from the passage tubes at 15, 30, and 75 days after being inoculated. These DNA extracts were submitted to the same PCR and DNA sequencing protocols described above.
Phylogenetic analysis
Partial sequences of the Borrelia fla gene generated in the present study were aligned by the ClustalX (Thompson et al. 1997) and manually refined by Genedoc (Nicholas et al. 1997) with corresponding B. burgdorferi s.l. sequences available in GenBank. The created alignment included 45 different sequences (408 bp). Sequences from Borrelia hermsii and Borrelia anserina were included as outgroups.
The 16S rRNA partial sequences of I. pararicinus generated in this study were aligned with corresponding sequences of 14 species of the I. ricinus complex available in GenBank, and published by Xu et al. (2003). The sequences of other Ixodes species known to occur in Uruguay, namely Ixodes auritulus, Ixodes loricatus, and Ixodes longiscutatus (Venzal et al. 2003), plus I. uriae (used as outgroup) were also included in the alignment.
Phylogenetic trees (Borrelia fla gene, and tick 16S rRNA) were inferred by the maximum parsimony methods and were performed with PAUP 4.0b10 software (Swofford 2002) with 1000 replicates of random-addition taxa and tree bisection and reconnection branch swapping; all positions were equally weighed. Bayesian analysis (tick 16S rRNA) was performed using MrBayes v3.1.2 (Ronquist and Huelsenbeck 2003). The tree searches employed GTR+GAMMA and proportion of invariable sites. The first 25% of the trees from 1,000,000 generations were discarded as burn-in.
Results
Among ethanol-preserved ticks, Borrelia DNA was detected by the fla-PCR in 2 (8.7%) out of 23 TC1 males, and 6 (54.5%) out of 11 TC2 males (Table 1). None of the 15 and 22 females of TC1 and TC2 ticks, respectively, contained Borrelia DNA. All these PCR-positive ticks yielded visible amplicons of the expected size in aragose gel by both the first and the nested reactions targeting the fla gene. DNA sequences of 459 to 617 bp were generated from the 8 PCR-positive male ticks; these sequences were shown to represent 5 different fla haplotypes (designated as A, B, C, D, E). Haplotypes A, B, and C differed from each other by only 1 to 2 single-nucleotide polymorphisms (SNPs) that resulted in no amino acid change. Similarly, haplotypes D and E differed from each other by only 2 SNPs that also resulted in no amino acid change. On the other hand, haplotypes A, B, and C differed from haplotypes D and E by 18–29 nucleotide substitutions that resulted in at least 4 amino acid changes. By BLAST analyses, haplotypes A, B, and C were most similar (99% identity) to corresponding fla sequences of B. bissettii DN127 (D82857, CP002746) and other B. burgdorferi s.l. sequences (AF264893–AF264897), whereas haplotypes D and E were most similar (97% identity) to corresponding fla sequences of B. burgdorferi s.s. (X69611, AE000783, AF264879–AF264881, AF264886, AF264889).
Phylogenetic analyses inferred from fla partial sequences showed that the 5 haplotypes (A–E) amplified from I. pararicinus belong to the B. burgdorferi s.l. group (Fig. 1). Haplotypes A–C formed a single cluster that grouped within the B. bissettii–B. carolinensis group under 100% bootstrap support, whereas haplotypes D and E segregated in a relatively distant cluster with B. americana, however with only 50% bootstrap support.
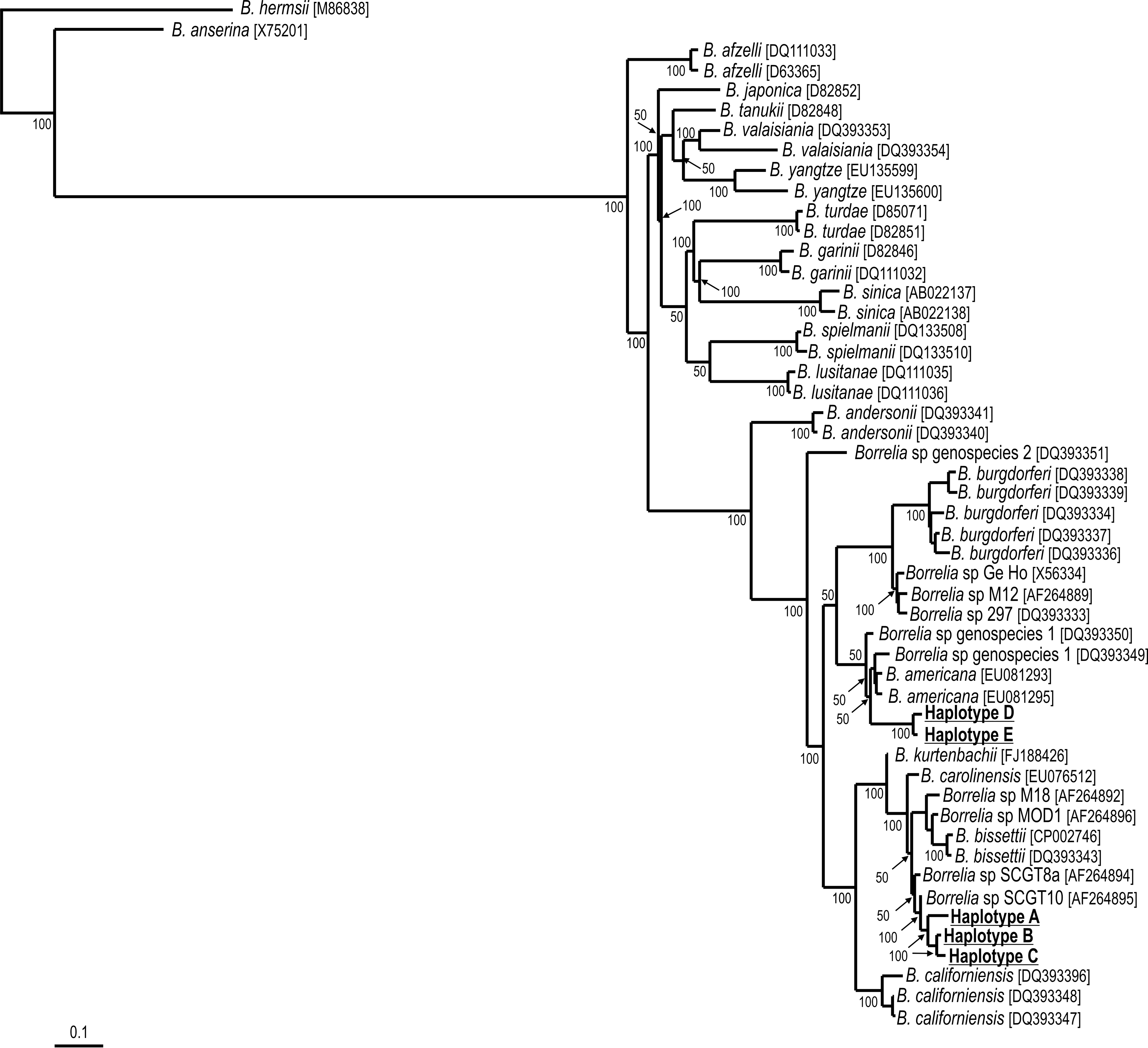
Maximum parsimony (MP) phylogenetic tree of Borrelia fla partial sequences from the B. burgdorferi sensu lato, and Borrelia haplotypes (A, B, C, D, E) from I. pararicinus ticks from Uruguay. The B. hermsii and B. anserina corresponding sequences were used as outgroups. Numbers at nodes are support values derived from bootstrap, 1000 replicates. Numbers in brackets are GenBank accession numbers.
Attempts to isolate Borrelia organisms in culture medium were performed with 54 TC3 adult ticks collected from cattle and 11 free-living TC4 nymphs. Weekly examinations of cultures by dark-field microscopy failed to demonstrate live Borrelia through the whole incubation period of 100 days. However, Borrelia DNA was detected by fla-PCR in culture media from 2 vials (1 inoculated with 5 TC3 males, and 1 inoculated with 5 TC4 nymphs) at 30, 45, and 90 days after inoculation. In addition, new culture tubes inoculated with passages of the 2 above PCR-positive cultures yielded Borrelia DNA at 15 and 30 days, but not at 75 days after being inoculated. DNA sequencing of the fla-PCR products revealed that the culture inoculated with 5 males contained B. burgdorferi s.l. haplotype A, whereas the culture inoculated with 5 nymphs contained B. burgdorferi s.l. haplotype B (100% identity with sequences of haplotypes A and B, respectively, detected in TC1 ticks) (Table 1).
Samples that generated Borrelia DNA by fla PCR also generated products by both the IGS- and the rrs-PCR protocols. DNA sequences for the IGS gene were generated from 3 TC2 ticks, whereas DNA sequences for the rrs gene were generated from the same 3 TC2 ticks, and from 1 PCR-positive culture. The 3 IGS sequences, designated as haplotypes F, M, and G, corresponded to the same ticks that generated fla haplotypes C, D, and E, respectively. While haplotypes M and G were very similar to each other (they differed by only 2 indels), they both differed by 11 nucleotides (94.3% similarity) from haplotype F. By BLAST analysis, haplotype F (195 bp) was most similar (99% identity) to the corresponding IGS sequences of Borrelia sp. SCW-30h (AF221673), and then 98% similar to Borrelia sp. SCGT-10 (AF221681) and B. bissettii (EF015627); haplotypes M (254 bp) and G (253 bp) were most similar (95–96% identity) to Borrelia sp. SCW-30a (HM802215) and B. americana (HM802219).
The 4 rrs sequences, designated as haplotypes I, J, K, and L corresponded to the same ticks that generated fla haplotypes B, C, D, and E, respectively. Similarly to fla analyses, these rrs sequences also formed 2 distinct groups, 1 composed by haplotypes I and J (100% similarity), and a second group composed by haplotypes K and L (99.9% similarity, differing each other by only a SNP). On the other hand, the rrs sequences of these groups (I and J versus K and L) were more polymorphic, with 98.7–99.3% similarities. By BLAST analysis, haplotypes I (343 bp) and J (706 bp) were closest (99–100% identity) to B. bissettii (CP002746) and Borrelia sp. strain Z41293 (AF091367), whereas haplotypes K (673 bp) and L (706 bp) were closest (99% identity) to multiple sequences of B. americana (HM802226, HM802225, EU081288–EU081288).
DNA sequences (410 bp) of a portion of the mitochondrial 16S rRNA gene were generated for 2 TC2 I. pararicinus ticks, corresponding to 2 males that were PCR-positive to Borrelia (1 of the males yielded fla haplotype C, the second male yielded fla haplotype D). Their 16S rRNA partial sequences were identical to each other, and by BLAST analysis were closest (95% similar) to corresponding sequences of I. affinis from Colombia (AF549861) and the United States (AF549834), and 93% similar to I. pararicinus from Argentina (AF549855). Phylogenetic analyses inferred from 16S rRNA partial sequences confirms that the I. pararicinus tick from Uruguay belongs to the I. ricinus complex (Fig. 2), as its sequence grouped with I. pararicinus from Argentina within a large cluster containing only species of this species complex, including the primary vectors of Lyme disease in the Northern Hemisphere, namely I. scapularis, I. pacificus, I. ricinus, and I. persulcatus.
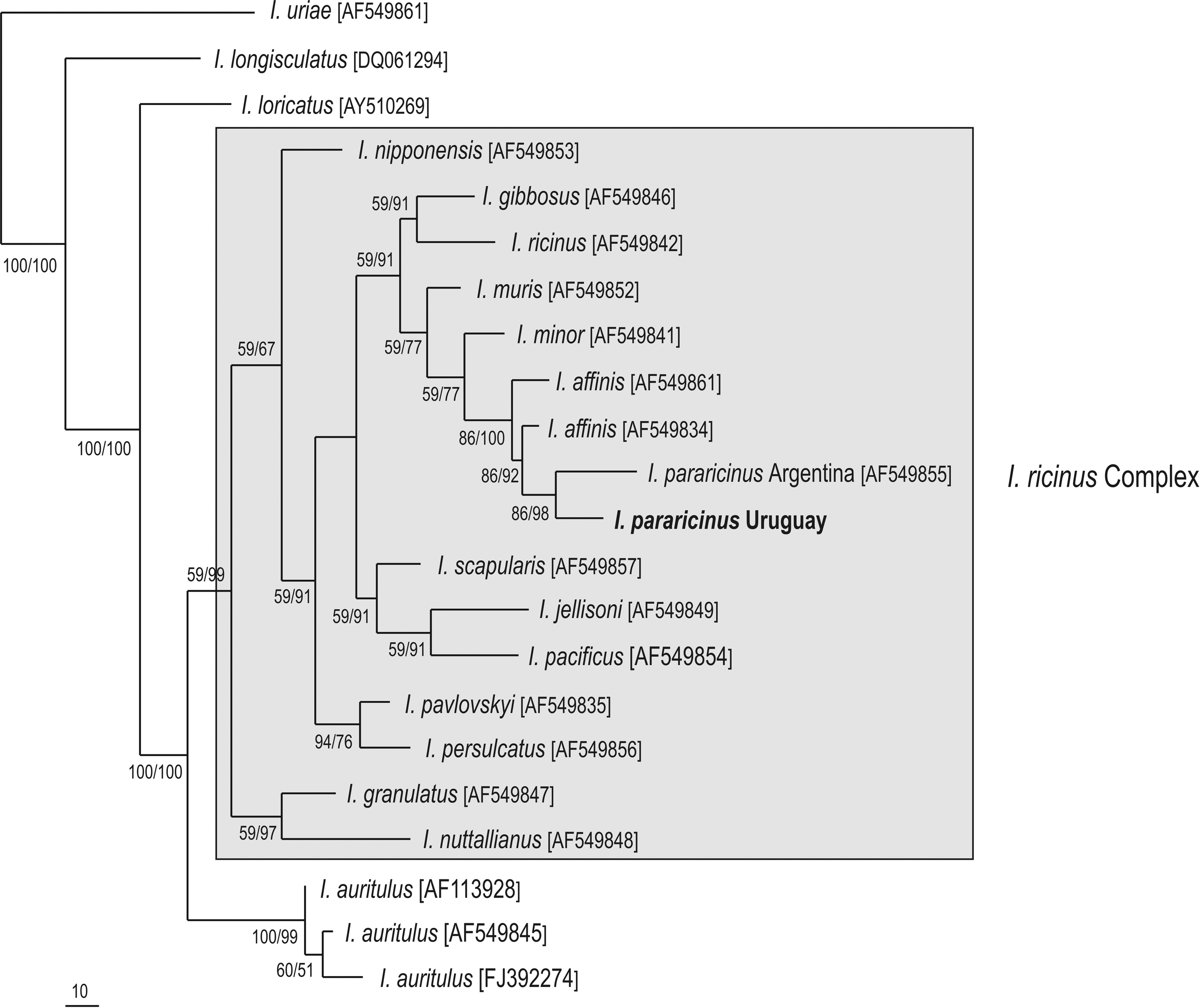
Maximum parsimony (MP) phylogenetic tree of 16S rDNA partial sequences of ticks from the I. ricinus complex, and Ixodes ticks from Uruguay. The I. uriae corresponding sequence was used as outgroup. Numbers at nodes are support values derived from bootstrap (1000 replicates for MP/ posteriori probability for Bayesian analysis). Numbers in brackets are GenBank accession numbers.
DNA sequences determined in this study have been deposited into GenBank and given the indicated accession numbers as follows: JX082311–JX082315 for the fla sequences, JX082316–JX082317 for the rrfA-rrlB IGS sequences, and JX082318–JX082321 for the rrs sequences of Borrelia haplotypes; JX082322 for the 16S rRNA sequence of I. pararicinus.
Discussion
In this study, different Borrelia haplotypes were found in ticks of the I. ricinus complex, namely I. pararicinus, from 2 different hosts and localities of Uruguay. Genetic analyses indicate that these haplotypes represent 2 different genospecies of the B. burgdorferi s.l. group; 1 genospecies, represented by fla haplotyes A, B, and C is phylogenetically associated with B. bissettii, whereas the other genospecies (represented by fla haplotyes D and E) is phylogenetically closest to B. americana. These results were reinforced by BLAST analyses of IGS and rrs sequences generated in this study.
Before the present study, the only report of B. burgdorferi s.l. infecting ticks of the Southern Hemisphere referred to B. garinii infecting the tick I. uriae infesting seabird colonies of Campbell Island (New Zealand) and Crozet Islands (Olsen et al. 1995, Gylfe et al. 1999). In addition, B. burgdorferi s.l., represented by at least 4 genotypes (including B. burgdorferi s.s.), have been reported infecting the tick I. auritulus from migratory birds in Canada (Morshed et al. 2005, Scott et al. 2010, 2012), which is relevant to the present study because I. auritulus has a nearly global distribution that includes Uruguay (Guglielmone et al. 2003). Indeed, the Borrelia haplotypes detected in the present study are clearly distinct from B. garinii (Fig. 1), and are not related to the genotypes previously reported infecting I. auritulus in Canada, because their IGS sequences (EU019109–EU019127) diverge 5–13% with the IGS sequences reported in the present study (data not shown). Therefore, even though I. auritulus and I. uriae are known to parasitize birds that migrate between the Northern and Southern Hemispheres, none of the currently known I. auritulus- or I. uriae-associated B. burgdorferi s.l. genotypes were found in the present study. These facts indicate that the Uruguayan B. burgdorferi genotypes circulate in a distinct enzootic cycle, yet to be elucidated. In fact, our phylogenetic analysis inferred from fla sequences suggests that the Uruguayan haplotypes potentially represent 2 geographically distinct new genospecies of the B. burgdorferi s.l., yet to be determined by successful culture and deeper molecular characterization.
Because we were able to detect spirochetal DNA in the cultures for as long as 90 days after inoculation, and for at least 30 days of the passages, we conclude that we isolated B. burgdorferi s.l. from I. pararicinus ticks in BSK medium. However, for unknown reasons, our isolates did not grow in a sufficient number to be visualized by dark-field microscopy, and therefore, did not establish in BSK medium. Because it is known that strains of B. burgdorferi s.l. differ in their capacity to grow well in culture (Stanek and Reiter 2011), it is possible that the Uruguayan Borrelia strains are more fastidious; therefore, different growth requirements should be further tested.
It is noteworthy that all I. pararicinus ticks found to be infected by B. burgdorferi s.l. were males or nymphs; no females were found to be infected, although this gender represented the majority of all tested ticks. Because it has been demonstrated in vitro that either deer or cattle serum complement has borreliacidal effects (Kurtenbach et al. 1998), and because the female ticks of the present study were all collected from deer or cattle, it is possible that strains of B. burgdorferi s.l. eventually present in female ticks before feeding were destroyed once the ticks fed on these hosts, therefore precluding a successful PCR detection or isolation. On the other hand, our detection of Borrelia in male ticks collected from deer or cattle is supported by the fact that Ixodes males rarely, if ever, feed on hosts under natural conditions, although they climb on hosts to copulate with feeding females (Sonenshine 1991).
Genetic analysis confirmed that the I. pararicinus ticks of the present study belong to the I. ricinus complex, corroborating our findings of B. burgdorferi s.l. in the only representative species of this tick complex in Uruguay. Because transovarial transmission of B. burgdorferi s.l. in Ixodes ticks is rare or nonexistent, the infection of nymphal and adult ticks depends on their feeding as larvae or nymphs on competent Borrelia-reservoir hosts, which are currently known to be different species of rodents and birds in the Northern Hemisphere (Stanek and Reiter 2011). Because both larvae and nymphs of I. pararicinus feed primarily on passerine birds and sigmodontinae rodents in Uruguay (Venzal et al. 2005), further studies are needed to test their reservoir competence for local isolates of B. burgdorferi s.l. Indeed, new ecologic studies are needed to determine the main vertebrate hosts associated with this I. pararicinus–B. burgdorferi s.l. interaction to elucidate the enzootic cycle and its potential to emerge as a new focus of Lyme borreliosis in South America.
Footnotes
Acknowledgments
We are grateful to Dr. Carlos G. de Souza, José Pedro Araujo, Fernando Ramos (Colonia Don Bosco-Laguna Negra), and Gustavo Casás for logistic support during field work. We thank park ranger Alejandro González (Parque Salus) for providing the deer. This work was supported by FAPESP, CNPq, CAPES (Brazil) and Programa 720- Contrapartida de Convenios (Uruguay-Brazil).
Author Disclosure Statement
Authors declare no conflict of interest.