
Abstract
Phlebotomine sand flies are known to transmit Leishmania, bacteria, and viruses that affect humans and animals in many countries worldwide. Sand fly–borne viruses belong mainly to the Phlebovirus, Vesiculovirus, and Orbivirus genera, and some of them are associated with outbreaks or sporadic human cases in the Mediterranean Europe. Up to now, Toscana virus is the only phlebovirus of medical importance identified in France. To study the diversity of the sand fly population living in the southeastern France, an entomological study was conducted from May to October, 2007. Most of the trapped sand flies belonged to Phlebotomus perniciosus (82.0%) and Sergentomyia minuta (17.3%) species; only three specimens were Phlebotomus ariasi. Molecular characterization of the P. perniciosus specimen based on the mitochondrial cytochrome b gene demonstrated different subpopulations living in the same areas. Most of the specimens belonged to the haplotypes pern01 and pern09, already described in France, but some belonged to original new haplotypes. The detection of one viral sequence clustering with Massilia/Granada virus, and of four sequences corresponding to two potential new phleboviruses (proposed names Olbia and Provencia viruses, respectively), revealed an unexpected diversity of phlebovirus species in France.
Introduction
Genetic analyses of Phlebotomus (Larroussius) perniciosus Newstead, a well-known vector of both Leishmania and phleboviruses in the western Mediterranean basin, showed this species complex contains few haplotypes (Pesson et al. 2004, Perrotey et al. 2005). A mitochondrial introgression with the closely related species Phlebotomus (L.) longicuspis was found and the P. (L.) perniciosus species was subdivided into three lineages—the widely spread lineage (Malta, Italy, France, Tunisia, and Morocco), which contains morphologically typical specimens; the Iberian lineage, also containing morphologically typical specimens; and the North African lineage (Morocco and Tunisia), containing morphologically atypical “P. (L.) longicuspis-like” males. The settlement in southern France and northeastern Spain, respectively, could be due to the expansion of segregate populations from southern Italy and Spain, respectively, during the Pleistocene Ice Age (Perrotey et al. 2005). The geographical limit between the widely spread lineage and the Iberian lineage and their possible hybridization, not yet described, may have occurred in southwestern France or in northern Spain (Perrotey et al. 2005). This gap highlights the need of a screening and isolation of viruses from individual sand flies in these areas where P. (L.) perniciosus is recorded. In France, typical specimens have ever been recorded. No atypical specimen has been emphasized.
To improve our knowledge about French sand flies, and particularly P. (L.) perniciosus, we studied sand flies trapped for a 6-month period in several locations near the Mediterranean coast in France. After species identification based on morphological features, P. (L.) perniciosus samples were investigated by molecular analyses to identify the haplotypes present in France.
Additionally, molecular screening of the samples allowed the detection of five phlebovirus sequences, four of which belonging to two putative novel lineages, tentatively named Olbia virus and Provencia virus.
Material and Methods
Specimen collection and treatment
Insects were collected alive from May through October, 2007, by using standard CDC miniature light traps (John W. Hock, Gainesville, FL). Collections took place in several locations between Marseilles and Toulon (Table 1), two French cities located on the Mediterranean coast and located ∼50 km to each other.
Sand flies were sorted from the collected insects and treated individually during all experimental steps. Main external morphological features were identified by microscopic observation. The head and genitalia of individual sand flies were cut off in a drop of ethanol using sterile needles, cleared in boiling Marc-André solution, and then mounted under a cover slip for identification (Depaquit et al. 2008). These slides are available upon request (J.D.).
The remaining tissues (thorax, wings, legs, and abdomen) were suspended in 400 μL of sterile phosphate-buffered saline, mixed with microbeads (Lysin Matrix E, MP Biomedicals, Life Science, France), and crushed at 40 vibrations per second in a tissue homogenizer (TissueLyser, Qiagen, France). Tissue lysates were stored at −80°C until use.
Sand fly molecular typing
The collected sand flies were molecularly typed based on the cytochrome b method (Esseghir et al. 1997). Tissue suspensions were centrifuged for 5 min at 12,000 g at 4°C. Total nucleic acids were extracted and purified from 200 μL of supernatant on a MagnaPure automate using a Total Nucleic Acids Isolation kit (Roche Diagnostics, Meylan, France) following the manufacturer's protocols. Amplicons were generated by PCR performed on a 50-μL volume using 5 μL of extracted DNA solution and 50 pmol of each of the two primers N1N-PDR and C3B-PDR following a previously published method (Esseghir et al. 1997) and then sequenced. Sequences were compared with sequences already published (Pesson et al. 2004, Depaquit et al. 2005, Perrotey et al. 2005) by using the Pregap and Gap software included in the Staden Package (Bonfield and Staden, 1996). Sequence alignment and neighbor-joining (NJ) trees were performed using the Kimura 2 parameters model included in the MEGA version 4 software (Tamura et al. 2007).
Viral molecular detection
Total nucleic acid extracts obtained as described above were used for RT-PCR detection of phleboviruses as matrix for phlebovirus genome amplification assays. All samples were processed individually using two methods: (1) The presence of the Toscana virus (TOSV) genome was assessed by a specific real-time RT-PCR (Perez-Ruiz et al. 2007) using the Superscript III Platinum One-Step qRT-PCR system; (2) the detection of phlebovirus RNA was done using a universal RT-nested PCR for phleboviruses and applied on nucleic acids extracts using degenerate primers that targeted a 166-nucleotide-long region within the L segment (Sanchez-Seco et al. 2003). Briefly, 10 μL of nucleic acids were submitted to reverse transcription (RT) using Superscript III RT (Invitrogen/life Technologies, Cergy Pontoise, France) according to the manufacturer's protocol, with the addition of 10% dimethyl sulfoxide in reverse transcription mix. Nested PCR was performed with Pfu DNA polymerase (Promega, France) in conditions recommended by the manufacturer in a final volume of 50 μL with 10 μ of cDNA and 5 μL of external amplicons.
Virus genome sequencing and analysis
PCR products were purified by ultrafiltration (Millipore, France) and sequenced on both strands using forward and reverse primers with Big Dye v.1.1 chemistry using ABI3730XL (Applied BioSystem). The obtained sequences were compared to phlebovirus sequences available in the GenBank database. NJ analyses were performed by using the Kimura 2 parameters model included in the MEGA version 4 software (Tamura et al. 2007). The robustness of the resulting tree was tested by 1000 bootstrap replications.
Results
Sand flies analysis
A total of 427 sand flies were captured from May to October, 2007, and were identified based on morphological characters: 82.0% belonged to P. (L.) perniciosus (254 males all showing the typical morphology and 96 females), 17.3% (31 males and 43 females) belonged to Sergentomyia minuta, and 0.7% (3 males) belonged to P. (L.) ariasi (Table 1). The sex ratio was 2/1 (67.4% males and 32.6% females).
The sequences of cytochrome b were determined for 94 P. perniciosus (Table 1). NJ analyses showed seven sand fly haplotypes of P. (L.) perniciosus that include morphologically exclusively typical specimens (Fig. 1). The haplotype pern01 already described in France (Perrotey et al. 2005) remained the predominant haplotype in our study (82 specimens) and five other specimens belonged to the haplotype pern09, also described previously in France (Perrotey et al. 2005). Some specimens defined five new haplotypes; four of them (PH157, PH277, PH275, and PH122/PH156) fell in the main branch that also included haplotypes pern01 and pern09, whereas PH383 defined a new divergent branch (Figs. 1 and 2). Among the specimens tested, none belonged to the Spanish haplotypes pern04 or pern05. GenBank accession numbers of sand flies sequenced are GU385219–GU385312.

Neighbor-joining tree based on 279 aligned base pairs of the cytochrome b mitochondrial DNA (mtDNA). The sequences from sand flies collected during this study (GenBank accession numbers GU385219–GU385312) were compared to Mediterranean haplotypes of P. perniciosus and P. longicuspis available in GenBank. The percent bootstrap values are indicated on the branches.

Alignment of mitochondrial cytochrome b related to the processed and published sand flies. Only the positions with variable nucleotides are featured. All the haplotypes correspond to typical P. perniciosus, except the haplotypes pern06 and pern07.
Molecular detection and identification of phlebovirus sequences
All of the 427 samples were investigated for the presence of phleboviruses by two RT-PCR assays. Negative results were obtained by using a TOSV-specific real-time RT-PCR. In contrast, a pan-phlebovirus RT-nested PCR (Sanchez-Seco et al. 2003) allowed the detection of virus sequences in five P. (L.) perniciosus haplotypes (PH131, PH225, PH320, PH373, and PH383), whereas no virus was detected in P. (L.) ariasi nor S. minuta. Interestingly, three virus sequences were found in male sand flies (Table 1).
In the studied genomic region, the sequence PH320 belonged to the Massilia/Granada group (bootstrap value of 99%), featuring with the other viruses of this group nucleotide and amino acid homologies >92.1% and >94.5%, respectively (Fig. 3).
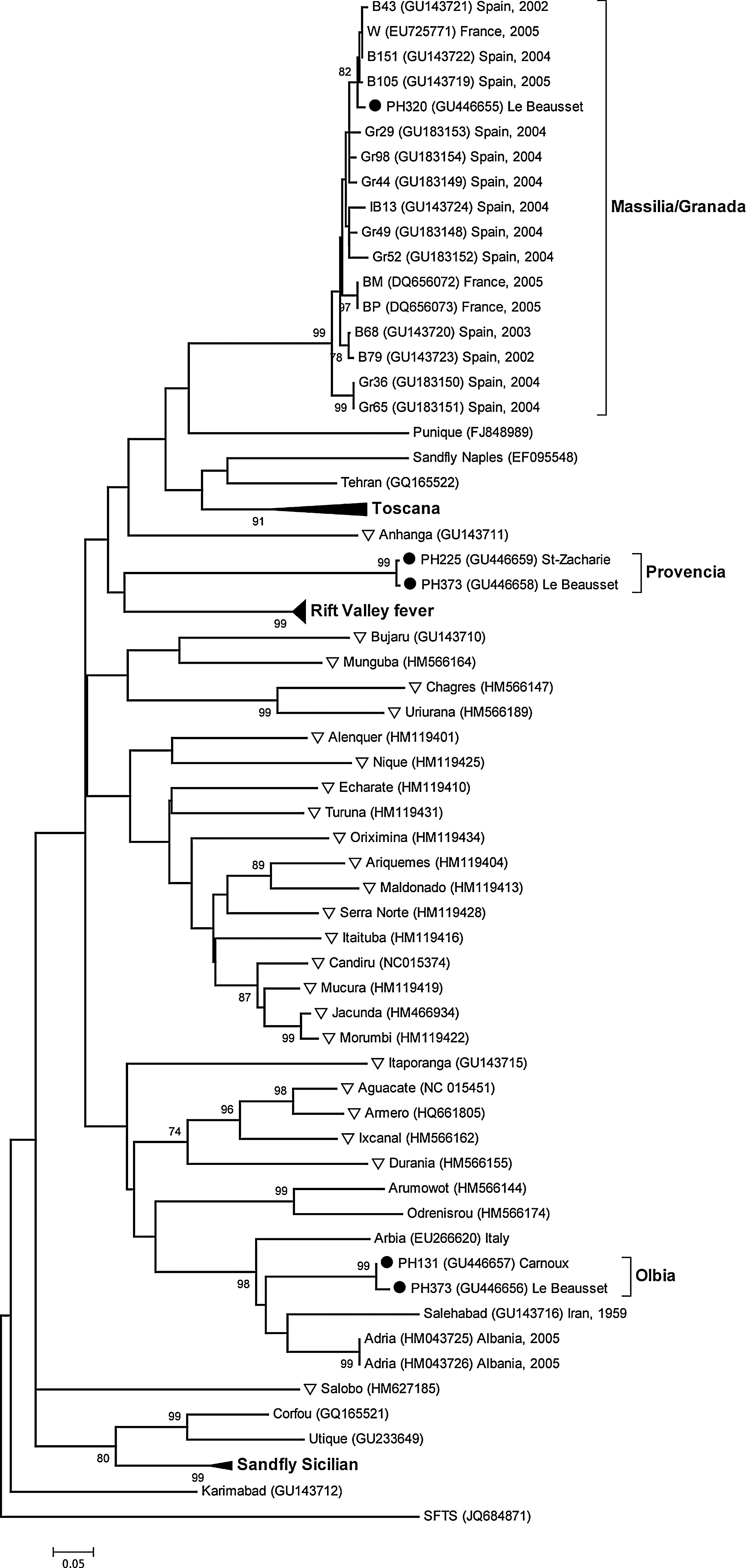
Dendogram based on 166-nucleotide-long sequences within the L segment. The sequences from the present study are indicated by full circles. New World phleboviruses are indicated by triangles. For better legibility of the tree, some lineages are collapsed and bootstrap values lower than 70 are not indicated. The respective location and year of isolation are indicated for some isolates of interest, if known.
The PH131 and PH373 sequences, found in sand flies collected at a distance of ∼20 km from each other, were very close to each other in the studied region (98.8% nucleotidic homology with two nucleotide divergences, 98.2% amino acid homology with one amino acid divergence). Phylogenetically, these two sequences clustered with Arbia, Salehabad, and Adria virus sequences (bootstrap value of 98%), three phleboviruses isolated in Italy, in Iran, and in Albania, respectively. Interestingly, PH131 and PH373 diverged noticeably from Arbia, Salehabad, and Adria virus sequences (Table 2) and were as divergent from these three virus sequences (nucleotide homology of 75.9–77.7%, amino acid homology of 85.5–89.1%) as these three virus sequences to each other (nucleotide homology of 74.1–79.5%, amino acid homology of 85.5–96.4%). Consequently, PH131 and PH373 were tentatively named Olbia, the name of a Greek colony established in the area in the 4th century B.C.E.
Nucleotide and amino acid homologies (%) are shown below and above the diagonal, respectively.
The PH225 and PH383 sequences, found in sand flies collected at a distance of ∼22 km from each other, were virtually identical in the studied region (only one nucleotide divergence and one amino acid divergence). These two sequences did not cluster with any other viruses, constituting together a new branch (Fig. 3). Among the other phleboviruses, Rift Valley fever virus (RVFV) displayed the highest degree of homology with these two sequences (nucleotide homologies of ∼60%, amino acid homologies of ∼56%) whereas all of the other virus sequences displayed a homology <58% and <51% at the nucleotide and amino acid levels, respectively. These results suggested that PH225 and PH383 belonged to a divergent phlebovirus lineage, tentatively named Provencia virus.
Attempts to isolate viruses in cell culture were unsuccessful, and no further genetic characterizations were conducted. GenBank accession numbers of virus sequenced are GU446655–GU446659.
Discussion
To improve our knowledge about the sand fly populations living in France, collections were organized in 2007, during the period when sand flies are active (Rioux et al. 1969, Charrel et al. 2009) and in a large diversity of sites whose altitudes ranged from 50 to 320 meters. Three different species were identified (P. perniciosus, P. ariasi, and S. minuta), with a majority of P. perniciosus (82%). This distribution was in agreement with the low altitude of the catching sites, P. (L.) ariasi being more abundant at higher altitudes. The species distribution and the sex ratio were also in agreement with a recent study conducted in the same area (Charrel et al. 2009).
Molecular analyses of genetic markers have improved the taxonomy of sand flies by allowing the discrimination of subpopulations that remain morphologically indistinguishable (Caterino et al. 2000). These markers can be used to study the phylogeographic patterns of sand fly populations and their respective spreading. Among these markers, the cytochrome b mitochondrial gene (Esseghir et al. 1997) has been widely used to depict the diversity of sand fly species in many countries bordering the Mediterranean Sea and in the Middle East (Esseghir et al. 2000, Aransay et al. 2003, Pesson et al. 2004, Hamarsheh et al. 2007, Moin-Vaziri et al. 2007, Parvizi et al. 2010, Belen et al. 2011, Dvorak et al. 2011). Performed on our specimens, cytochrome b analysis revealed the predominance within the French P. perniciosus population of the haplotype pern01 (88%), already described in France. Nonetheless, an unexpected diversity of haplotypes was observed; five other haplotypes were detected, four for the first time.
Phleboviruses are endemic in the Mediterranean region of Europe and could spread to more temperate areas where vectors are abundant, emphasizing the importance to carry out survey studies in the European Union (Konstantinou et al. 2007). Therefore, phlebovirus detection assays were conducted on our samples.
Until now, TOSV is the only phlebovirus whose medical importance has been demonstrated in France (Peyrefitte et al. 2005, De Lamballerie et al. 2007, Bichaud et al. 2011). The two major genotypes of TOSV (Spanish and Italian) were documented close to Marseilles (Charrel et al. 2007, Collao et al. 2009), and viral TOSV RNA was detected in the same geographic region in S. minuta, which is not known as a usual TOSV vector (Charrel et al. 2006). No TOSV was detected in our samples by using a TOSV-specific RT-PCR assay. These negative results confirm the low frequency of detection of this virus in sand flies trapped in this area (Charrel et al. 2007), despite the high anti-TOSV antibody prevalence observed in humans (De Lamballerie et al. 2007). In fact, 12% of sera from blood donors and 18.9% of sera from patients hospitalized for central nervous system (CNS) infection contained immunoglobulin G (IgG) reacting against TOSV or TOSV-related phleboviruses (De Lamballerie et al. 2007).
Five phlebovirus sequences were detected by using a generic RT-PCR assay targeting the polymerase gene. Despite their short length, the corresponding amplicons can be used for virus identification after sequencing (Sanchez-Seco et al. 2003).
One sequence was found very close to virus sequences already described in France and Spain (nucleotide homology of more than 92.1%, amino acid homology of more than 94.5%) and constituting together the Massilia/Granada group (Charrel et al. 2009, Collao et al. 2010). This group is made of viruses isolated in the 2000s from sand flies trapped in France, in mainland Spain, and in the Balearic Islands (Charrel et al. 2009, Collao et al. 2010). Massilia isolates and Granada ones are closely related viruses based on the L and S segments, but they diverge noticeably in the M segment (Collao et al. 2010).
The four other sequences did not belong clearly to any cluster already described. While related to Arbia, Salehabad, and Adria viruses, PH373 and PH131 constituted a separate branch in the studied region, tentatively called Olbia virus. Even more significantly, PH225 and PH383 constituted a novel phylogenetic cluster, tentatively called Provencia virus, that displayed a nucleotide homology of less than 60% with all the other known phleboviruses. These new types will have to be confirmed in the future by resolving larger sequences in the three genomic segments of the corresponding isolates.
Compared to previous studies, our approach based on individual specimen analysis allowed an accurate estimation of sand fly infestation. The overall phlebovirus sequence prevalence observed in the present study within P. (L.) perniciosus was 1.43% (2.08% in females and 1.18% in males), in accordance with the prevalence already observed in other studies, i.e., from 0.22% through 0.75% when sand flies were individually tested and from 4.2% through 9.1% when tested in pools containing up to 30 insects (Charrel et al. 2009). Interestingly, three of our sequences were found in male sand flies, suggesting vertical transmission of these phleboviruses (Tesh and Modi 1987). However, venereal transmission of viruses from female to male is possible during copulation and cannot be excluded in the present study. Thus, males could be involved in the maintenance of the phleboviruses in the insect population by venereal contamination of the females.
In conclusion, our study uncovered the genetic diversity of sand flies in southeastern France and highlighted the circulation among these insects of several phlebovirus lineages, some of which had never been described before. Because infected sand flies were mainly captured in villages and suburban areas, the transmission of these phleboviruses to humans by sand flies bites, the impact of these viruses on human health, and their potential to give rise to human outbreaks will have to be evaluated in the near future, considering the continued urban expansion into the woodland of southern France.
Footnotes
Acknowledgments
This work was conducted as part of the Service de Santé des Armées, the Direction Générale pour l'Armement (DGA), the Institut Pasteur (IP), and the Institut de Veille Sanitaire (IVS St-Maurice). The authors thank Dr. Jon Davis for reviewing the manuscript.
Author Disclosure Statement
The contents of this publication are the responsibility of the authors and do not necessarily reflect the views of the French Armed Forces, the DGA, the IP, or the IVS.