
Abstract
Differential gene expression by Borrelia burgdorferi spirochetes during mammalian infection facilitates their dissemination as well as immune evasion. Modulation of gene transcription in response to host immunity has been documented with the outer surface protein C, but the influence of transcription of other genes is largely unknown. A low-density array (LDA) was developed to study transcriptional activity of 43 B. burgdorferi genes and 19 host genes that may be involved in various host–agent interactions. Gene transcription in heart, joint, and muscle tissue was compared in immunocompetent C3H and immunodeficient C3H-scid mice during early (3 weeks) and late (2 months) B. burgdorferi infection. Among all tissue types, levels of relative transcription of over 80% of B. burgdorferi genes tested were one- to nine-fold less in C3H mice compared to C3H-scid mice. At the later time point, all genes were transcribed in C3H-scid mice, whereas transcription of 16 genes out of 43 tested was not detected in analyzed tissues of C3H mice. Our data suggest that during infection of immunocompetent mice, a majority of B. burgdorferi genes tested are downregulated in response to acquired host immunity. LDA revealed variable patterns of host gene expression in different tissues and at different intervals in infected mice. Higher levels of relative expression for IL-10 during both early and late infection were detected in heart base, and it was unchanged in the tibiotarsal joint. Comparative analysis of B. burgdorferi and host genes transcriptional activity revealed that increased flaB mRNA during early infection was followed by increases of CCL7, CCL8, interleukin-10 (IL-10), and tumor necrosis factor-α (TNF-α) in all assessed tissue types. LDA represents a valuable approach for sensitive and quantitative gene transcription profiling and for understanding Lyme borreliosis.
Introduction
Numerous studies have proposed several possible mechanisms of B. burgdorferi immune evasion, including binding and invasion of ECM and connective tissue (Liang et al. 2004a, Zambrano et al. 2004), antigenic variation (Bankhead and Chaconas 2007, Palmer et al. 2009), suppression of harmful immune responses (Diterich et al. 2003, Hovis et al. 2006, Bhide et al. 2009), and selective expression of surface antigens (Liang et al. 2002a, Liang et al. 2004a, 2004b). Lipidated spirochetal outer surface proteins are recognized by Toll-like receptor 2 (TLR2) (Lien et al. 1999), which leads to activation of monocytes and neutrophils, with subsequent secretion of inflammatory cytokines (Lazarus et al. 2006, Strle et al. 2009, Shin et al. 2010).
Host immune pressure and associated spirochetal immune evasion are dynamic processes, so that many genes are likely to be variably expressed as a consequence of host immunity. In addition, expression of surface antigens may confer tissue specificity and guide dissemination within the host and may therefore be differentially expressed in various tissues and different stages of infection. For example, several ligand-binding lipoproteins, including BmpA outer surface protein and its three paralogous proteins, BmpB, BmpC, and BmpD, play a role in infection by binding to laminin (Verma et al. 2009), and it has been shown that bmpA- or bmpB-deleted mutants are unable to persist in mouse joint tissues (Pal et al. 2008). Decorin-binding protein (Dbp) A and B bind to tissues rich in decorin, including joints, heart base, and skin. These collagen-rich sites seem to favor persistence of spirochetes with higher dbpA expression (Liang et al. 2004a). Deletion of dbpBA impedes dissemination of spirochetes and alters long-term survival (Weening et al. 2008). Other ECM ligand-binding outer surface proteins include fibronectin-binding protein (Fbp), Borrelia glycosaminoglycan-binding protein (Bgp), RevA, ErpX, P66 (αIIbβ3 integrin), and an unidentified type I collagen-binding adhesion, among others (Probert and Johnson 1998, Defoe and Coburn 2001, Parveen et al. 2006, Cabello et al. 2007, Brissette et al. 2009a, b).
Pathogen profiling of B. burgdorferi obtained by techniques such as serotyping (Wilske et al. 1996), immunohistochemistry (Miklossy et al. 2008), multilocus enzyme electrophoresis (Boerlin et al. 1992), in situ hybridization (Fuchs et al. 1992), ribotyping (Postic et al. 1996), pulsed-field gel electrophoresis (Belfaiza et al. 1993), plasmid fingerprinting (Xu and Johnson 1995), randomly amplified polymorphic DNA (Wang et al. 1998), species-specific PCR and PCR-based restriction fragment length polymorphism (RFLP) analysis (Le Fleche et al. 1997, Marconi and Garon 1992), and sequence analysis (Postic et al. 1998) have all been used to help understand the pathogenesis of Lyme borreliosis, but transcriptional activity of selected genes is becoming an essential part of such analysis. Several new molecular techniques such as membrane-based array (Hyde et al. 2006), and microarray (Adusumilli et al. 2010), show promise in that they are objective, quantitative, robust, and allow high throughput.
Advances in gene quantification technologies allow quantitative, sensitive, and robust expression profiling of multiple genes in single samples simultaneously. One such technology, low-density array (LDA), is a medium-throughput method based on a quantitative real-time reverse transcription PCR (qRT-PCR) platform (Abruzzo et al. 2005). LDA allows the simultaneous measurement of expression of up to 384 genes in a single sample with minimal handling of the samples, thereby decreasing contamination and operator-induced errors and allowing standardization of method and data interpretation across laboratories (de Cremoux et al. 2004).
Despite these recent advances in gene transcription detection technology, there has been limited transcriptional analysis of large clusters of B. burgdorferi genes (Anderton et al. 2004, Tokarz et al. 2004, Hyde et al. 2006), especially in the infected host (Liang et al. 2002b). In an initial survey to understand the global effect of host immune response on gene transcription of B. burgdorferi during infection in the mouse model, expression profiles of 43 different genes were selected, including those presumed to be involved in attachment, cell envelope, metabolism, complement regulation, cellular processes, and replication. We used a qRT-PCR-based method on the LDA platform to compare transcriptional levels of selected B. burgdorferi genes and 19 mouse chemokines and cytokines in mice during early (3 weeks) and late (2 months) infection.
Materials and Methods
Mice
Specific pathogen-free, 3- to 5-week
Borrelia burgdorferi
A clonal strain of B. burgdorferi sensu stricto (cN40) was grown in modified Barbour–Stoenner–Kelly (BSK) II medium (Barbour 1984). Spirochetes were grown to mid-log phase, harvested, and resuspended to 105 cells/mL in BSK II medium. Eight C3H mice and eight C3H-scid mice were infected by intradermal inoculation at the dorsal thoracic midline with 104 spirochetes each and then randomly divided into groups of four mice each. Eight C3H mice were sham-inoculated with BSK II medium and randomly divided into two equal groups. At 3 weeks and 2 months after inoculation, four infected C3H mice, four infected C3H-scid mice, and four sham-inoculated C3H mice were necropsied at each time point. To confirm infection, the urinary bladder and the inoculation site were cultured from each mouse, as described previously (Barthold et al. 1993).
Tissue collection and nucleic acid isolation
Heart base, ventricular myocardium, quadriceps muscle, and tibiotarsal joint were collected for analysis. Samples for DNA and cDNA analysis were immediately weighed, snap-frozen in liquid nitrogen, and stored at −80°C before nucleic acid extraction. For isolation of template DNA for quantitative analysis of spirochetes and total RNA to assess transcriptional activity of selected genes, we used ∼10 mg of tissue, as been described previously (Hodzic et al. 2003).
DNA amplification
For quantitative analysis of DNA isolated from tissue samples, flaB qPCR was performed as previously described (Hodzic et al. 2002). Absolute sensitivity and reproducibility of the qPCR was established by limiting dilution assay of cultured spirochetes and of positive tissue samples. The analytical sensitivity was in the range of 1–109 spirochetes, with a yield of detection close to 90% of the calculated amount of known target in each sample. Specificity of the qPCR was confirmed by the lack of amplification from negative control samples, and amplification was confirmed from positive control samples.
cDNA synthesis
For transcriptional activity analysis of the selected B. burgdorferi and host genes, first-strand cDNA was synthesized from total RNA using the QuantiTect Reverse Transcription Kit (Qiagen) in 50-μL reactions, according to the manufacturer's instructions. Because limited numbers of spirochetes and spirochetal RNA (cDNA) in tissue samples restricted the analytical sensitivity, the Advantage® 2 Polymerase Mix (Clontech Laboratories) was used to increase fidelity, efficiency, and greater yield of cDNA, according to the manufacturer's instructions. The preamplification reaction involved activation at 95°C for 15 sec, amplification for 25 cycles at 95°C for 15 sec, 55°C for 15 sec, and 70°C for 45 sec, followed by elongation at 70°C for 5 min. The preamplified products were diluted at a ratio of 1:10 and used as templates for qPCR analysis. All samples were analyzed for the presence of 18S rRNA to determine the efficiency of the nucleic acid extraction, amplification, and as an indicator of inhibition.
LDA assays
LDA assays were designed on the microfluidic card (Applied Biosystems) that contains eight sample-loading ports, each connected by a microchannel to 48 miniature reaction chambers for a total of 384 wells per card. A total of 43 B. burgdorferi N40 genes were selected from the National Center for Biotechnology Information (NCBI) GenBank (Schutzer et al. 2011). Genes were selected from different locations throughout the B. burgdorferi N40 genome (chromosome, linear plasmids, and circular plasmids), and among 12 different categories according to their putative functions. The selected genes encode proteins with a variety of functions, including attachment, cell envelope, motility, cell division, metabolism, cellular processes (toxin production, resistance, adaptation, response), DNA metabolism, complement regulation, persistence, general functions, protein synthesis, and hypothetical (Table 1).
Locked nucleic acid (LNA) probe (Roche, Pleasanton, CA).
Another LDA was designed to assess transcriptional activity of host genes, including MyD88 (5′-AGCTGCTGGCCT TGTTAGACC, 3′- TCTGGCAGTCCTCCTCGATG, probe #17 from Roche, Pleasanton, CA), cytokines interleukin-1β (IL-1β; Life Technologies, Grand Island, NY), IL-2 (Life Technologies), IL-4 (Life Technologies), IL-6 (5′-TTCACAAGTCGGAGGCTTAATTACA, 3′-AAGTGCAT CATCGTTGTTCATACA, probe FAM- TGAGAAAAGAGTT GTGCAATGGCAATTCTG), IL-10 (Life technologies), IL-12p40 (Life Technologies), IL-17α 5′ACTCCCTTG GCGCAAAAGT, 3′-AGGGTCTTCATTGCGGTGG, probe #50 from Roche), IL-21 (Life Technologies), interferon-γ (IFN-γ) (5′-TGAATAACTATTTTAACTCAAGTGGCATAG, 3′-ATA ATCTGGCTCTGCAGGATTTTC, probe FAM- TCTTGGATATCTGGAGGAACTGGCAAAAGG), tumor necrosis factor-α (TNF-α) (5′-TGGCCTCCCTCTCATCAGTT, 3′-GCTACAGGCTTGTCACTCGAATT, probe FAM-CCCAGACCCTCAC ACTCAGATCATCTTCT), and chemokines CCL2 (5′-ACCA GCAAGATGATCCCAATG, 3′-GAGCTTGGTGACAAAAACTACAGC, probe #19 from Roche), CCL7 (5′- GATCTCTGCCACGCTTCTGTG, 3′-GCATTGGGCCCATCTGG, probe #89 from Roche), CCL8 (5′-CAACATGAAGATCTACGCAG TGC, 3′-AGCCTTATCTGGCCCAGTCA, probe #26 from Roche), CCL12 (5′- ACCCCAGTCACGTGCTGTTAT, 3′-TGGTCCTGAAGATCACAGCTTC, probe #2 from Roche), CXCL12 (Life Technologies), CXCL13 (Life Technologies), CCL19 (5′-GGGTGCTAATGATGCGGAAG, 3′-GGTGAACACAACAGCAGGCA, probe # 17 from Roche), and CCL21 (Life Technologies). Gene-specific primers and hydrolysis probes were embedded in each well. The synthesized cDNA template from each sample (30 μL) was added to 50 μL of 2×Universal PCR Master Mix (Applied Biosystems) in a 100-μL reaction mixture. The mixture was added to each line of the microfluidic card after vortexing and brief centrifugation. The system allows analysis of four different samples at the same time, in duplicate. The PCR amplifications were performed in the microfluidic card sample block of an ABI Prism 7900HT sequence detection system (Applied Biosystems). The amplification was done under the following thermal cycle conditions: 2 min at 50°C to 10 min at 94.5°C, followed by 40 cycles of denaturation at 97°C for 30 sec, and annealing and extension at 59.7°C for 1 min.
For each of 43 B. burgdorferi N40 and 19 mouse target genes, two specific primers and one internal, fluorescence-labeled probe were designed with Primer Express software (Applied Biosystems). The amplification efficiency (E) of all assays was calculated from the slope of a standard curve generated on a 10-fold dilution in triplicate for every cDNA sample using the formula E=10(−1/slope) − 1. To obtain accurate and reproducible results, all assays were determined to have an efficiency of >95%. On the basis of the amplification efficiencies, detection limits were approximately 10 copies of cDNA per reaction. The coefficient of variability of the qPCR determined for 10 replicates was 15% or less. The specificity of each assay was determined using DNA or cDNA templates in duplicate from uninfected mouse tissues and other pathogens (Leptospira spp., Escherichia coli, Listeria monocytogenes, Staphylococcus aureus, Helicobacter spp.).
Relative quantification of transcriptional activity of each target gene was calculated using the equation 2−ΔΔCq. The method uses the transcriptional activity of a target gene based on E and Cq deviation (ΔCq) of tissue samples from infected C3H mice versus same sample types obtained from infected C3H-scid mice, and expressed in comparison to a reference gene (Pfaffl 2001). Cq values of target genes of all samples were normalized to a reference gene (16S rRNA) to compensate sample-to-sample and run-to-run variations and to ensure experimental reliability. Additional calculation was performed using Relative Expression Software Tool (REST
The stability of six potential B. burgdorferi reference genes (16S rRNA, flaB, gapDH, p13, p66, dbpA) was assessed between individual mice and mouse groups, between different tissue samples (heart, quadriceps muscle, and tibiotarsus), and at the different time points by using BestKeeper software (
Pathology
Knee and tibiotarsal joint (the other knee and tibiotarsal joint were used for nucleic acid extraction) were fixed in 10% neutral-buffered formalin, decalcified, paraffin embedded, sectioned, and stained with Hematoxylin & Eosin using standard technique. Slides were examined blindly for presence of arthritis, and severity of inflammation was scored on a scale of 0 (negative) to 3 (severe), as previously described (Barthold et al. 2006).
Statistical analysis
All statistical analyses were conducted with PASW Statistics 18 (IBM). To evaluate the transcriptional level differences of each target gene between tissues, heart base, ventricular muscle, tibiotarsal joint, quadriceps musle during early (3 weeks), and late (2 months) infection in C3H compared to immunodeficient C3H-scid mice or infected C3H compared to uninfected C3H mice, one-way anlaysis of variance (ANOVA) followed by a least-squares difference post hoc test was performed. The significance level for this test was considered p<0.05. The transcriptional activity ratio of genes was also tested by REST
Results
Infection status
All C3H and C3H-scid mice inoculated with B. burgdorferi cN40 were confirmed to be infected by culture (all urinary bladders and all inoculation sites positive). None of the sham-inoculated mice was culture-positive. B. burgdorferi flaB DNA was detected in all tissues of all infected mice, but not in sham-inoculated mice. The total bacterial loads, based on flaB DNA copy numbers, in heart base, ventricular muscle, tibiotarsal joint, and quadriceps muscle, were significantly higher in C3H-scid mice compared to C3H mice at 3 weeks and 2 months after inoculation (Fig. 1, p<0.05). The mean bacterial loads in tissues of both C3H and C3H-scid mice decreased over time (3 weeks compared to 2 months), but not significantly (p=0.08, p=0.26, respectively).
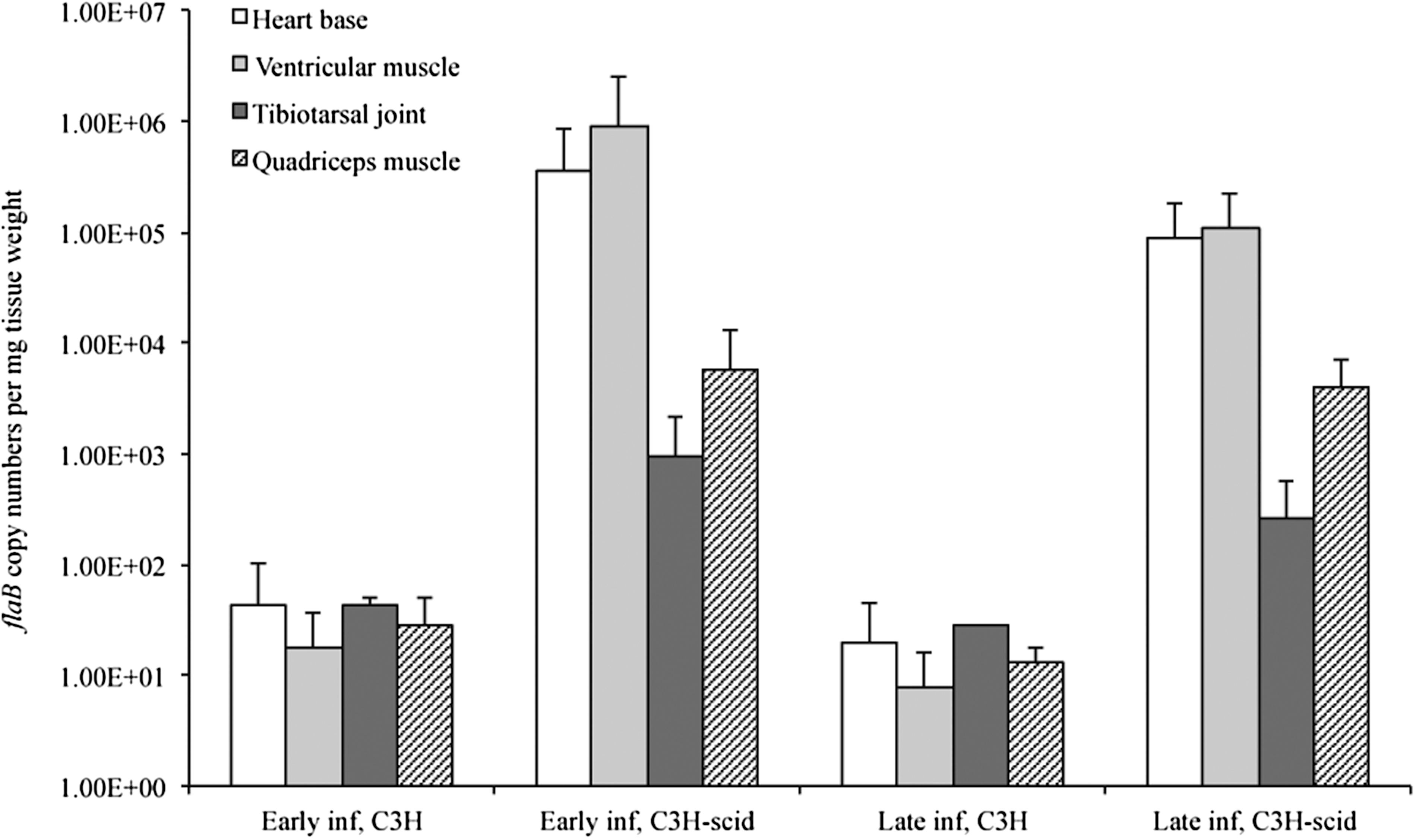
Copy numbers of flaB DNA (mean±standard deviation [SD]) in heart base, ventricular muscle, tibiotarsal joint, and quadriceps muscle of immunocompetent C3H and immunodeficient C3H-scid mice during early (3 weeks) and late (2 months) infection.
Histology
C3H-scid mice developed tibiotarsal arthritis (2+ severity at 3 weeks and 3+ severity at 2 months). Inflammation was mild (1+) in only one of four tibiotarsal joints of C3H mice at 3 weeks and absent in C3H mouse joints at 2 months. Knee inflammation was not scored, but present in all C3H-scid mice at both intervals, and in none of the infected C3H mice. Heart base, which is consistently inflamed during early infection (Armstrong et al. 1992), was used for DNA and cDNA extraction, and therefore was not available for histology.
LDA analysis
To determine the reproducibility of the LDA assay, five different dilutions of B. burgdorferi culture, as well as mouse tissues, were analyzed for all B. burgdorferi and host target genes. Intra-assay variations were found to be 1.6–3.2%, and interassay variations of the same sample in three separate runs were 4.1–5.3%. The data also demonstrated that using the LDA assay, it was possible to accurately detect two-fold changes for every target gene, and small changes could be detected at both low Cq (high expression) and high Cq (low expression). Total RNA was also extracted from all tissue samples from sham-inoculated mice and was subjected to RT-qPCR. Transcriptional activity was detected for host 18S rRNA, but not for any of the B. burgdorferi genes.
Selection of reference genes
The Cq values were plotted to compare transcriptional activity of 16S rRNA, flaB, gapDH, p13, p66, and dbpA of B. burgdorferi (Fig. 2). Transcription of dbpA mRNA was detected in all tissue samples obtained from both C3H and C3H-scid mice during early and late infection. Therefore, this gene was included as a potential reference gene for normalization against the other reference genes, as suggested by Bustin et al. (2010). The evaluated genes had different patterns of transcription within the same sample type, different sample types, and different mouse groups. Genes with higher coefficients of variation (CV) included p66 (17.08%), p13 (23.62%), and gapDH (33.34%), and were thus characterized as the least stable genes. The most stable reference gene identified with high transcriptional activity and invariant of the condition studied was 16S rRNA (8.33%), followed by flaB (15.24%) and dbpA (16.05%). A good indicator of constant RNA transcription over all tissue samples is a transcriptional range. The lowest range was observed for 16S rRNA (7.59), followed by p66 (9.72), flaB (9.92), p13 (12.88), dbpA (13.61), and gapDH (14.96). The reference gene candidates evaluated for transcriptional stability for normalization of mouse gene transcription included 18S rRNA, Actβ, B2M, and gapDH. The most stable host reference genes detected for this study were 18S rRNA and gapDH.
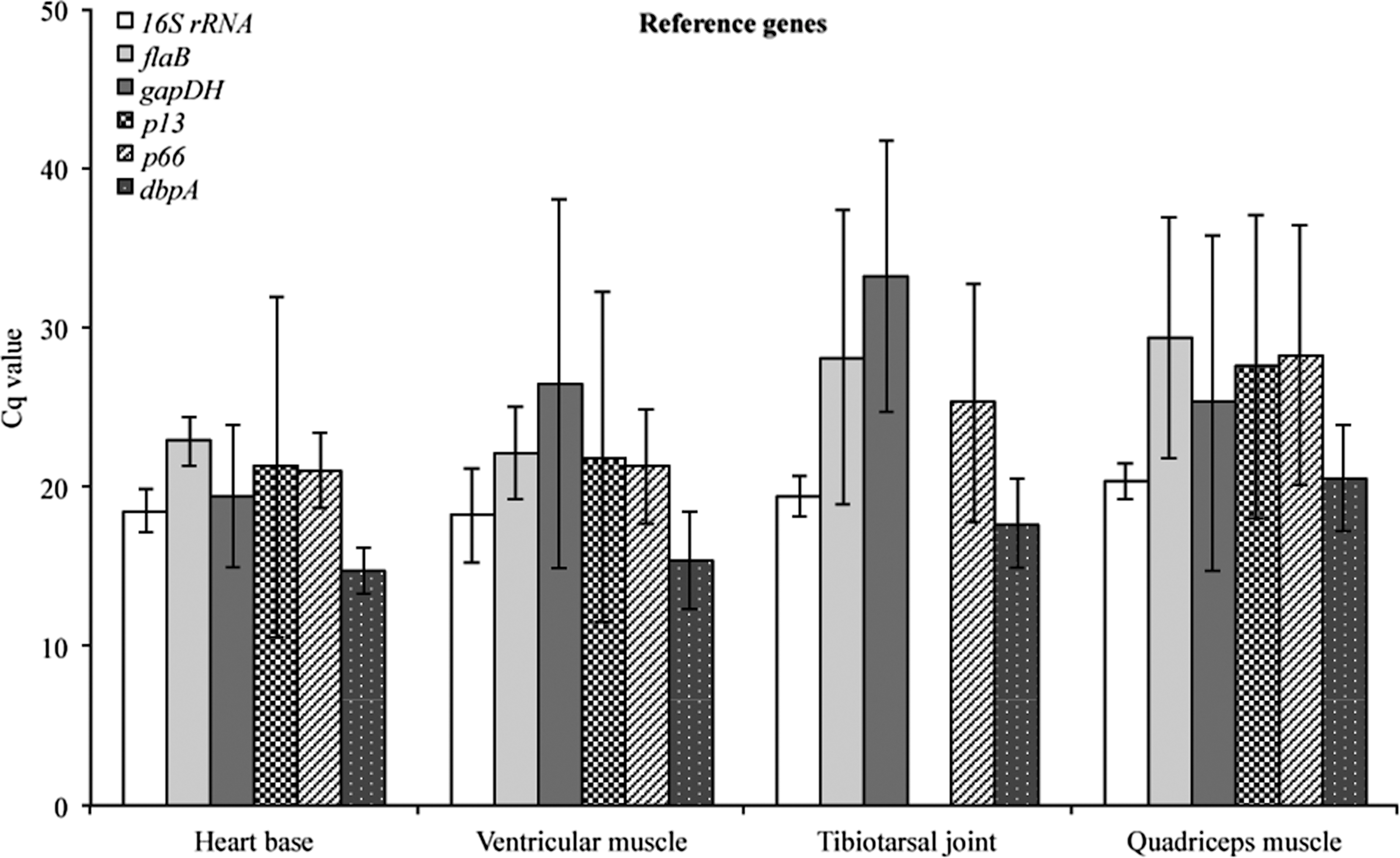
Validation of transcriptional mRNA stability of 16S rRNA, flaB, gapDH, p13, p66, and dbpA as candidates for selecting reference genes. Absolute Cq values (mean±standard deviation [SD]) were plotted within the same sample type, different sample types, and different mouse groups during early and late infection.
Transcriptional activity of B. burgdorferi
LDA analysis of heart base, ventricular muscle, tibiotarsal joint, and quadriceps muscle revealed that all analyzed B. burgdorferi genes were transciptionally active in all early and late infected C3H-scid mice (data not shown). In contrast to C3H-scid mice, B. burgdorferi in C3H mice did not transcribe detectable amounts of ospA, relA/spoT, oms38, acrB, and ackA mRNA during either early or late infection. Transcription was not detected for vlsE, eppA, bptA, crasp1, glpA, gapDH, S2, alr, colV, and p23-T2 at 2 months, whereas BB0142 transcription was not detected at 3 weeks of infection in C3H mice. During early infection of C3H mice, ospC transcription was detected in nine of 12 tissues at early infection, but only in one of 11 tissues at the later infection interval. As expected, ospA transcription was not detected in any of the C3H mouse samples, but low levels of ospA transcription were detected in C3H-scid mice (3/16 during early infection and 7/12 during late infection)(data not shown).
The spirochetal response to host immunity was complex. Transcriptional activity of the analyzed B. burgdorferi genes was significantly (p<0.05) higher in C3H-scid mice compared to C3H mice at each time point (data not shown). Comparing transcription of genes within each mouse group related to time of infection, no significant differences were found for C3H or C3H-scid mice (p=0.08 and p=0.11, respectively). However, at the 2-month interval of infection, transcription of several genes was downregulated compared to early infection in C3H mice.
Tissue samples from infected C3H and C3H-scid mice were analyzed for a relative B. burgdorferi transcriptional activity profile between the two types of mice to compare the global effects of the immune response on B. burgdorferi gene expression. Cq values obtained from every gene and all tissue types were normalized to 16S rRNA, and transcription level differences of ΔCq values were calculated between tissues of C3H versus C3H-scid mouse samples. Analysis showed higher levels of transcription for the majority of assessed genes in immunodeficient compared to immunocompetent mice. The transcription of B. burgdorferi genes that had changes (up- and/or downregulation) in tissues of C3H mice compared to C3H-scid were plotted and depicted (Figs. 3 –6). Levels of relative expression for over 80% of genes were one- to nine-fold lower in C3H mice compared to C3H-scid mice, or no signal was detected. During early infection, only fbp and flaB were upregulated in heart base of C3H mice compared to C3H-scid mice (Fig. 3), whereas eight genes involved in attachment (bgp, dbpA), motility (flaB), DNA metabolism (recA), and complement regulation (p21, erp22, erp23, erp27) were upregulated in ventricular muscle (Fig. 4). In the tibiotarsal joint, flaB, tgt, recA, and erp27 were upregulated during early infection (Fig. 5), whereas in quadriceps muscle, flaB, p66, S2, and ospE were upregulated (Fig. 6). The results indicated that B. burgdorferi in tibiotarsal joints and quadriceps muscle transcribed fewer genes compared to heart base and ventricular muscle during both early and late infection in C3H mice.
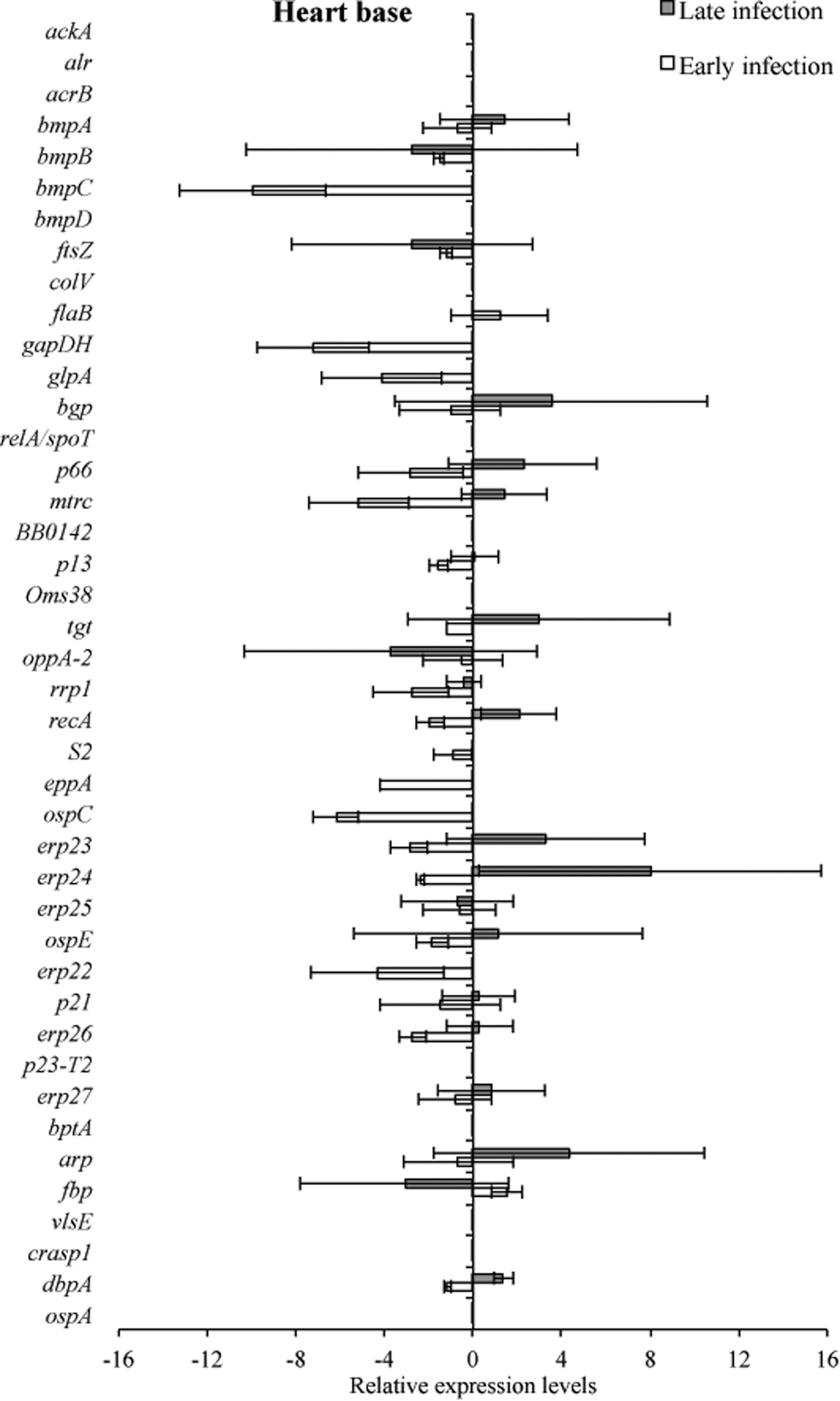
RNA transcription levels (means±standard deviation [SD]) of 42 B. burgdorferi genes in the heart base of C3H mice relative to gene transcription levels in C3H-scid mice during early and late infection. Samples from C3H mice were normalized to samples from C3H-scid mice, expressed in comparison to a reference gene (16S rRNA), and plotted as fold-changes for each gene. Bars to the left of the center line indicate reduced relative expression and bars to the right indicate increased relative expression in C3H mice compared to C3H-scid mice. Zero relative expression indicates no expression in C3H mice.
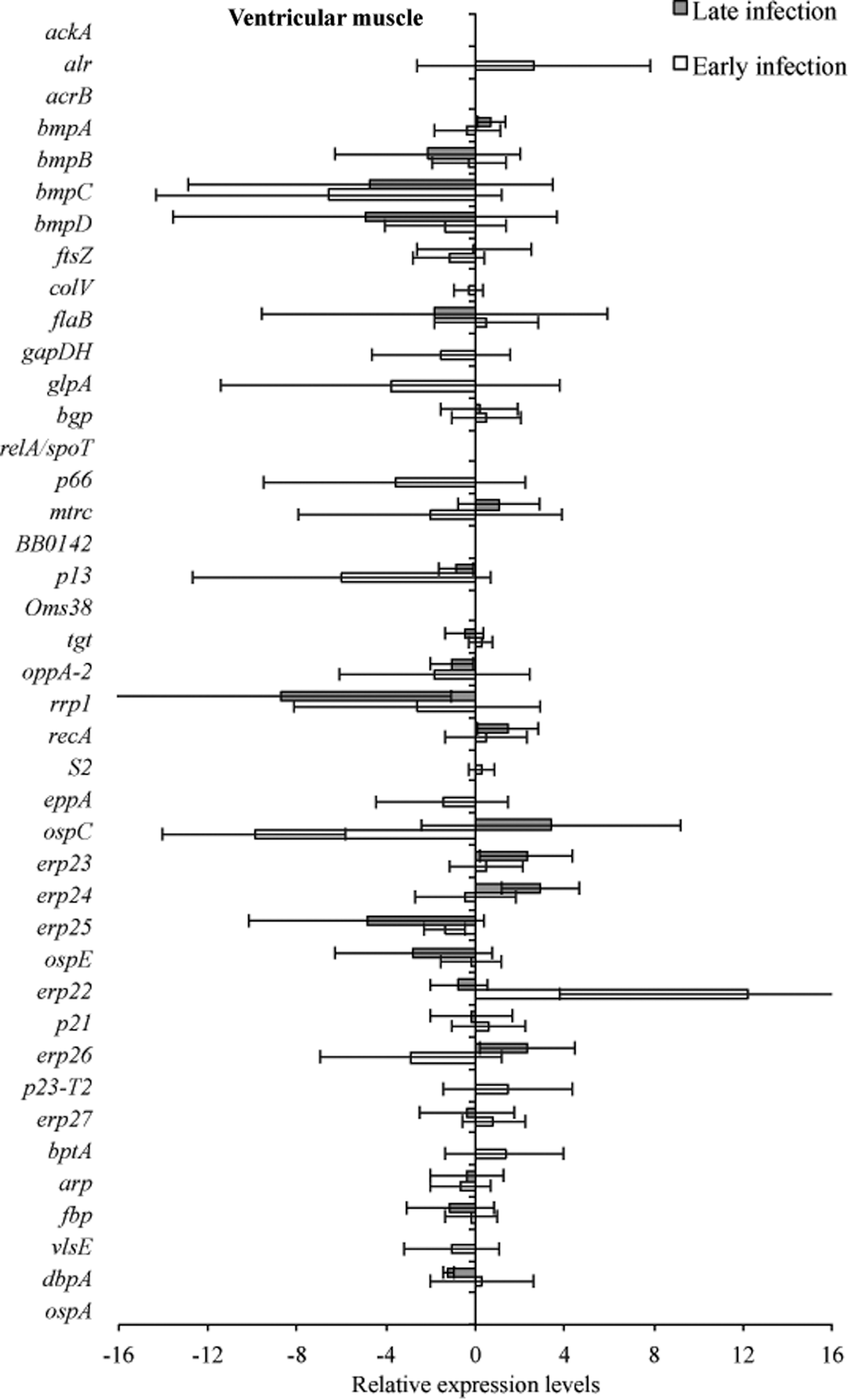
RNA transcription levels (means±standard deviation [SD]) of 41 B. burgdorferi genes in ventricular muscle of C3H mice relative to gene transcription levels in C3H-scid mice during early and late infection. Samples from C3H mice were normalized to samples from C3H-scid mice, expressed in comparison to a reference gene (16S rRNA), and plotted as fold changes for each gene. Bars to the left of the center line indicate reduced relative expression and bars to the right indicate increased relative expression in C3H mice compared to C3H-scid mice. Zero relative expression indicates no expression in C3H mice. Transcriptional activity was not detected for crasp1 in C3H or C3H-scid mice during early or late infection, and was therefore not plotted.
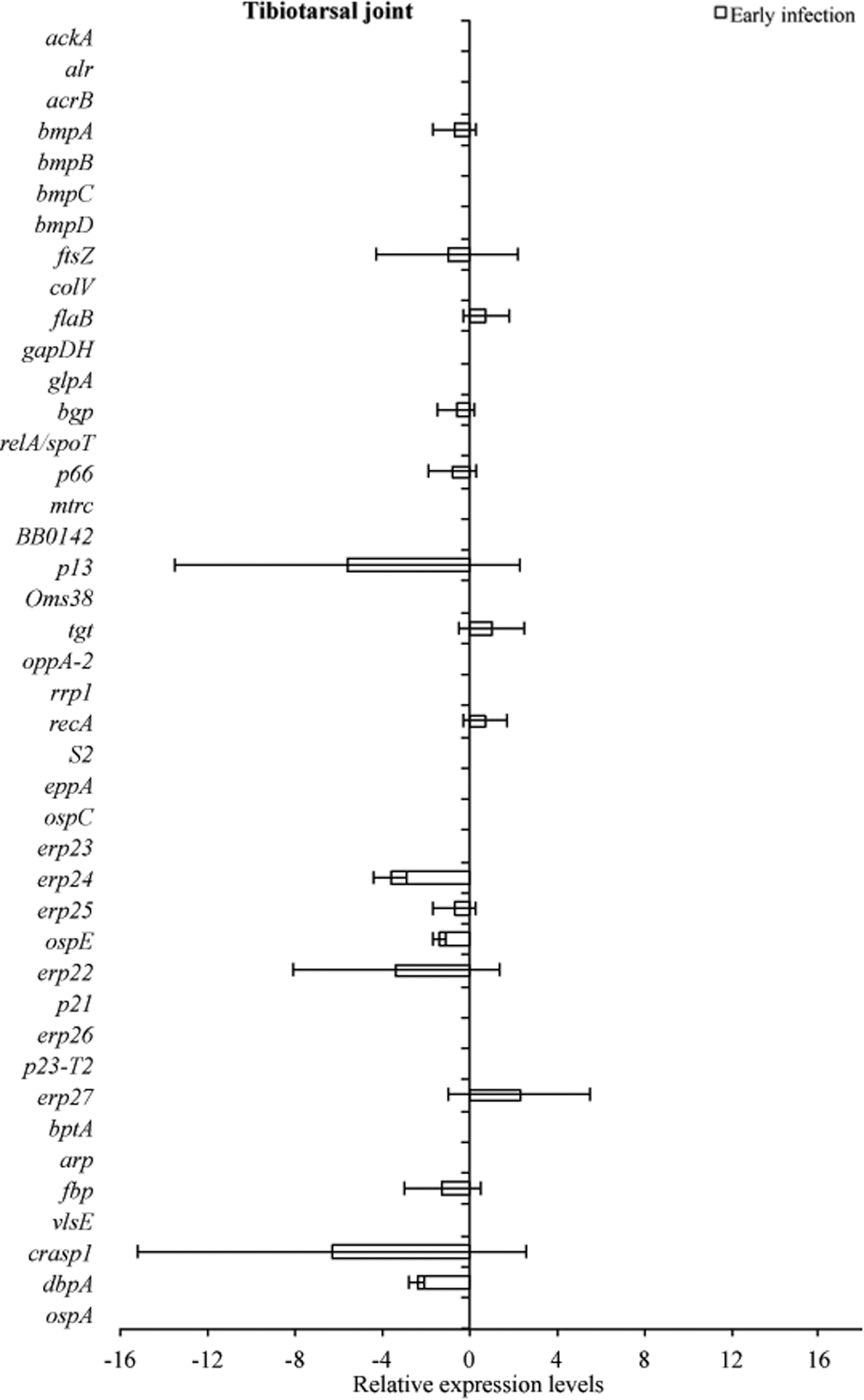
RNA transcriptional levels (mean±standard deviation [SD]) of 42 B. burgdorferi genes in tibiotarsal joint of C3H mice relative to gene transcriptional levels in C3H-scid mice during early infection. Samples from C3H mice were normalized to samples from C3H-scid mice, expressed in comparison to a reference gene (16S rRNA), and plotted as fold-changes for each gene. Bars to the left of the center line indicate reduced relative expression and bars to the right indicate increased relative expression in C3H mice compared to C3H-scid mice. Zero relative expression indicates no expression in C3H mice. B. burgdorferi mRNA was not detected in tibiotarsal joint samples collected at late infection, so the transcriptional activity was not assessed.
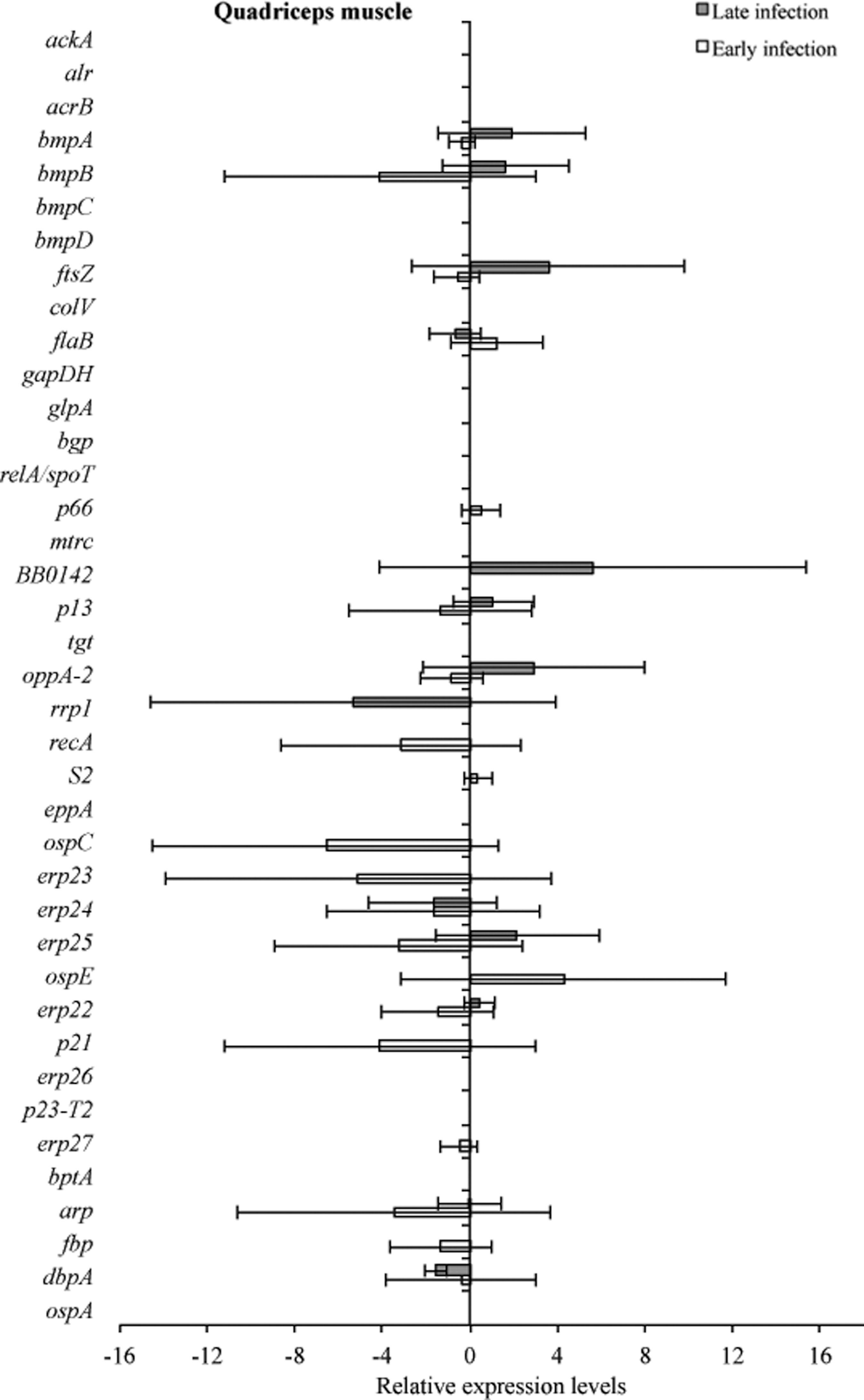
RNA transcriptional levels (mean±standard deviation [SD]) of 39 B. burgdorferi genes in the quadriceps muscle of C3H mice relative to gene transcriptional levels in C3H-scid mice during early infection. Samples from C3H mice were normalized to samples from C3H-scid mice, expressed in comparison to a reference gene (16S rRNA), and plotted as fold changes for each gene. Bars to the left of the center line indicate reduced relative expression and bars to the right indicate increased relative expression in C3H mice compared to C3H-scid mice. Zero relative expression indicates no expression in C3H mice. Transcriptional activity was not detected for crasp1, vlsE, and oms38 genes in both infected C3H and C3H-scid mice during early and late infection, and was therefore not plotted.
At 2 months after inoculation, transcription of 16 genes was not detected in analyzed tissues of C3H mice (Figs. 3 –6). Among 29 genes with detectable transcription, 14 were upregulated (one- to seven-fold increase) in heart base, including genes involved in attachment (bgp, dbpA, p66), cell envelope (ospE, bmpA, p13), DNA metabolism (recA), complement regulation (p21, erp23, erp24, erp26, erp27), cellular processes (mtrc), and unknown function (arp)(Fig. 3). Fewer genes were upregulated in ventricular muscle of C3H mice, including genes involved in attachment (bgp), cell envelope (bmpA), DNA metabolism (recA), complement regulation (erp23, erp24, erp26), and cellular processes (mtrc) (Fig. 4). In quadriceps muscle at late infection, transcription was detected for 13 genes, of which 7 (oppA-2, ftsZ, bmpA, bmpB, p13, erp22, erp25) were upregulated in C3H mice compared to C3H-scid mice (Fig. 6). Due to inadvertent loss of samples, only one tibiotarsal joint at later infection (positive for flaB DNA) was available for LDA analysis, but transcription was not detected for any of 43 genes. Interestingly, the ftsZ gene, which has been shown to play role in cell division of B. burgdorferi (Dubytska et al. 2006), was downregulated in all samples during both early and late infection in C3H mice compared to C3H-scid mice.
In summary, all genes were transcriptionally active in C3H-scid mice in all tissues. In contrast, the majority of B. burgdorferi genes were downregulated in C3H mice relative to C3H-scid mice, with upregulation of various genes, depending upon tissue type and interval of infection.
Transcriptional activity of host genes
To compare transcription of host immunoregulatory genes in response to infection, gene transcription levels were compared between infected C3H mice relative to uninfected C3H mice at 3 weeks and 2 months of infection. Comparative analysis of the heart base and ventricular muscle during early infection revealed similar significantly (p<0.05) higher levels of relative expression for CCL7, CXCL13, IL-10, and TNF-α, but not significantly CCL8 (p=0.15), whereas IL4 was downregulated in ventricular muscle (Figs. 7 and 8). At the later infection interval, more cytokines and chemokines were upregulated compared to early infection. During late infection, IL-2, IL-12p40, and TNF-α were downregulated in heart base, while IL-4 (p=0.07) and CXCL12 (p<0.05) were downregulated in ventricular muscle.
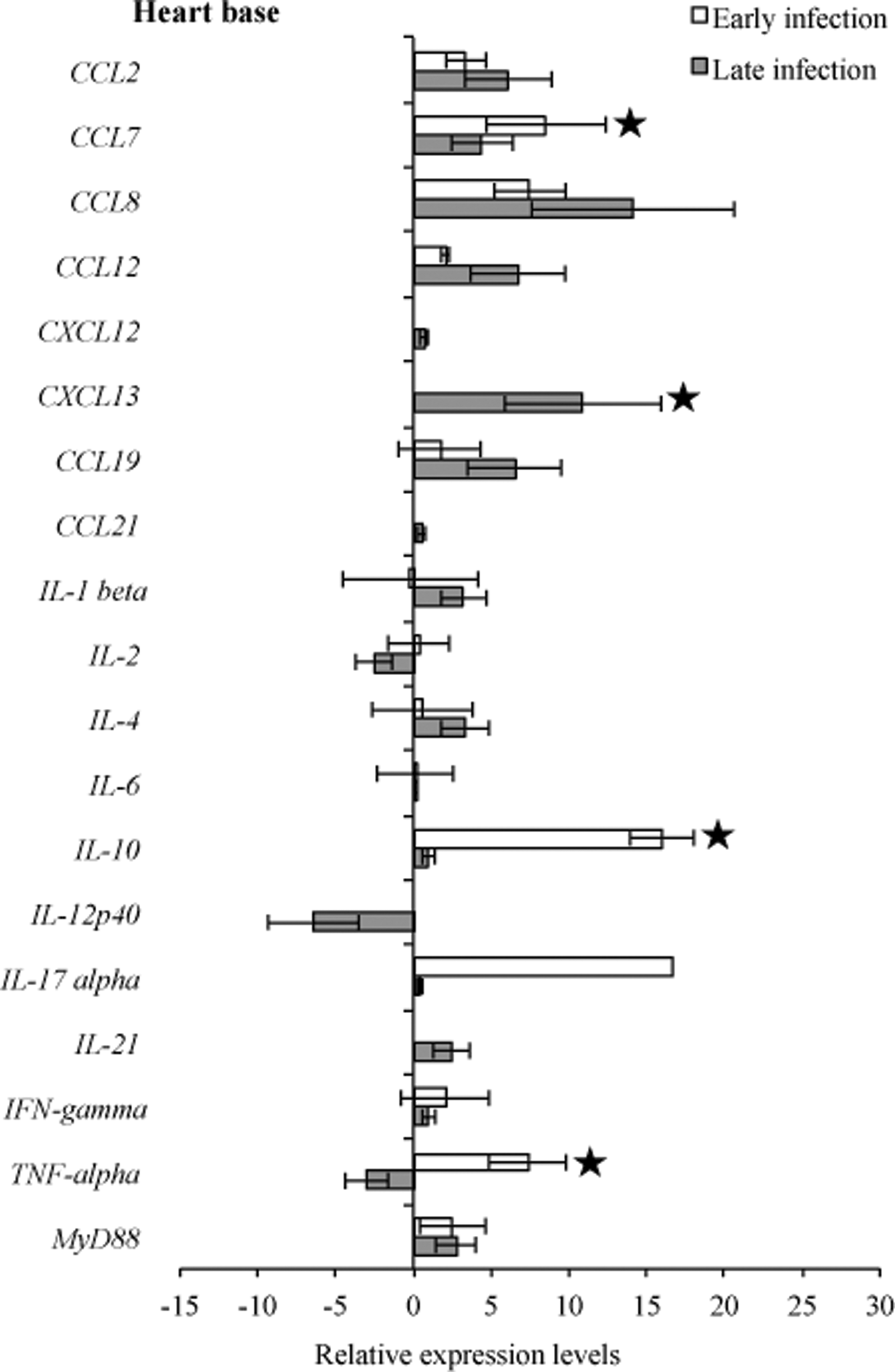
RNA transcription levels (means±standard deviation [SD]) of 19 mouse genes in the heart base of infected C3H mice relative to gene transcription levels in uninfected (sham inoculated) C3H mice during early and late infection. Samples from infected C3H mice were normalized to samples from sham-inoculated mice, expressed in comparison to reference genes (18S rRNA, Actb, B2M, and gapDH), and plotted as fold changes for each gene. Bars to the left of the center line indicate reduced relative expression and bars to the right indicate increased relative expression in infected C3H mice compared to sham-inoculated mice. Zero relative expression indicates no expression in infected C3H mice. Mouse genes not assessed in mice during early infection were CXCL12, CXCL13, CCL21, and IL-21.
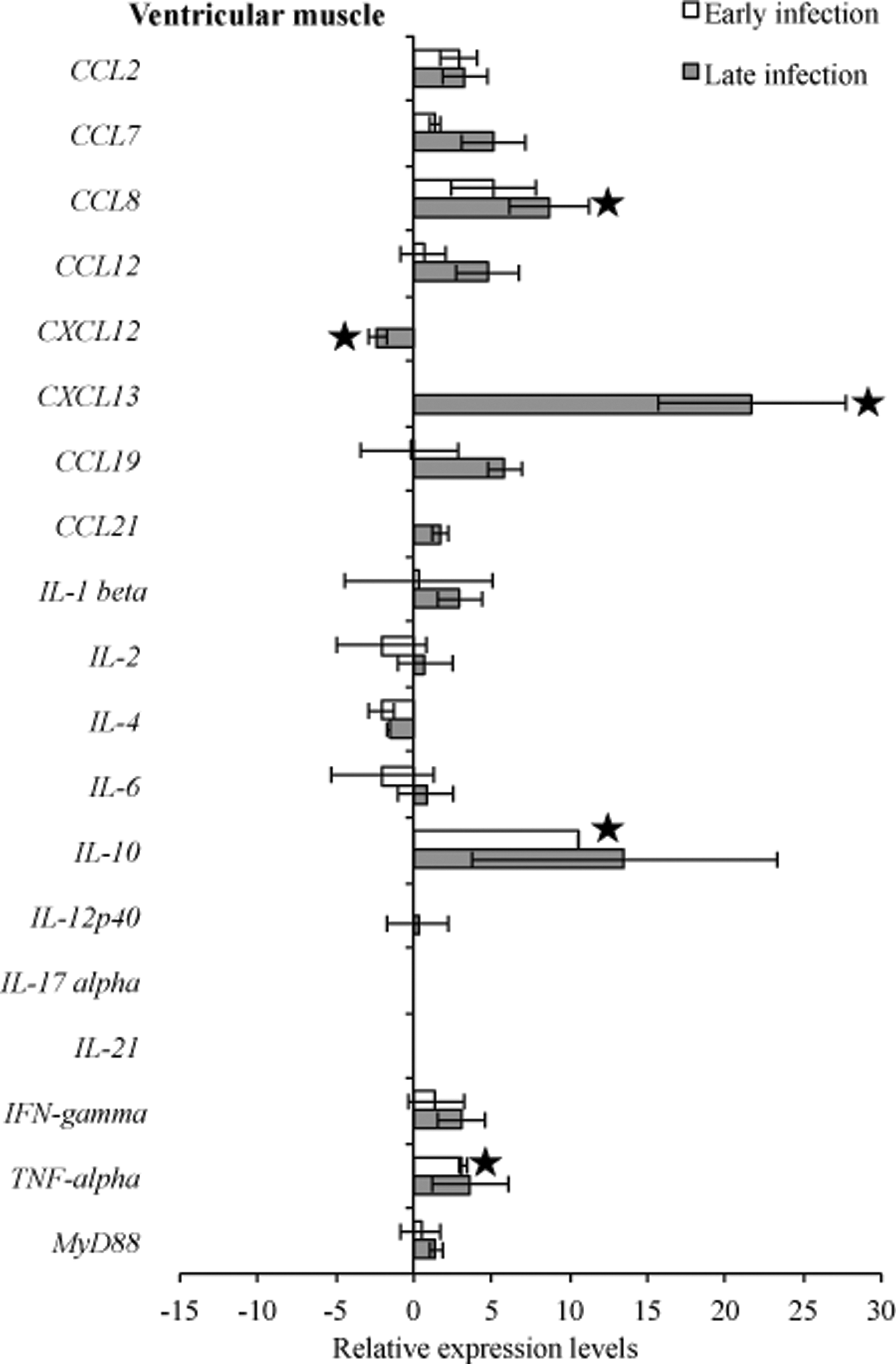
RNA transcription levels (means±standard deviation [SD]) of 19 mouse genes in the ventricular muscle of infected C3H mice relative to gene transcription levels in uninfected (sham inoculated) C3H mice during early and late infection. Samples from infected C3H mice were normalized to samples from sham-inoculated mice, expressed in comparison to reference genes (18S rRNA, Actb, B2M, and gapDH), and plotted as fold changes for each gene. Bars to the left of the center line indicate reduced relative expression and bars to the right indicate increased relative expression in infected C3H mice compared to sham-inoculated mice. Zero relative expression indicates no expression in infected C3H mice. Mouse genes not assessed in mice during early infection were CXCL12, CXCL13, CCL21, and IL-21.
Detectable levels of mouse target mRNA during both early and late infection in tibiotarsal joint and quadriceps muscle were fewer (12 samples for both tissues) compared to heart base and ventricular muscle (19 and 17 samples, respectively) (Figs. 9 and 10). In the tibiotarsal joint during early infection, CCL2, CCL7, and TNF-α were upregulated, while CCL12, CCL19, and MyD88 were downregulated, but not significantly. At late infection in the tibiotarsal joint, only CXCL13 was upregulated (p<0.05), while CCL2, CCL7, CCL8, CCL12, CXCL12, CCL19, CCL21, IL-1β, IL-10, TNF-α, and MyD88 were downregulated (Fig. 9). Figure 10 depicts transcriptional activity of mouse genes in quadriceps muscle during early and late infection. During early infection, all detected cytokines were upregulated, while at late infection CCL2, CCL7, CCL8, CXCL12, CXCL13, CCL19, IL-10, and TNF-α were relatively upregulated and CCL12 and MyD88 were downregulated.
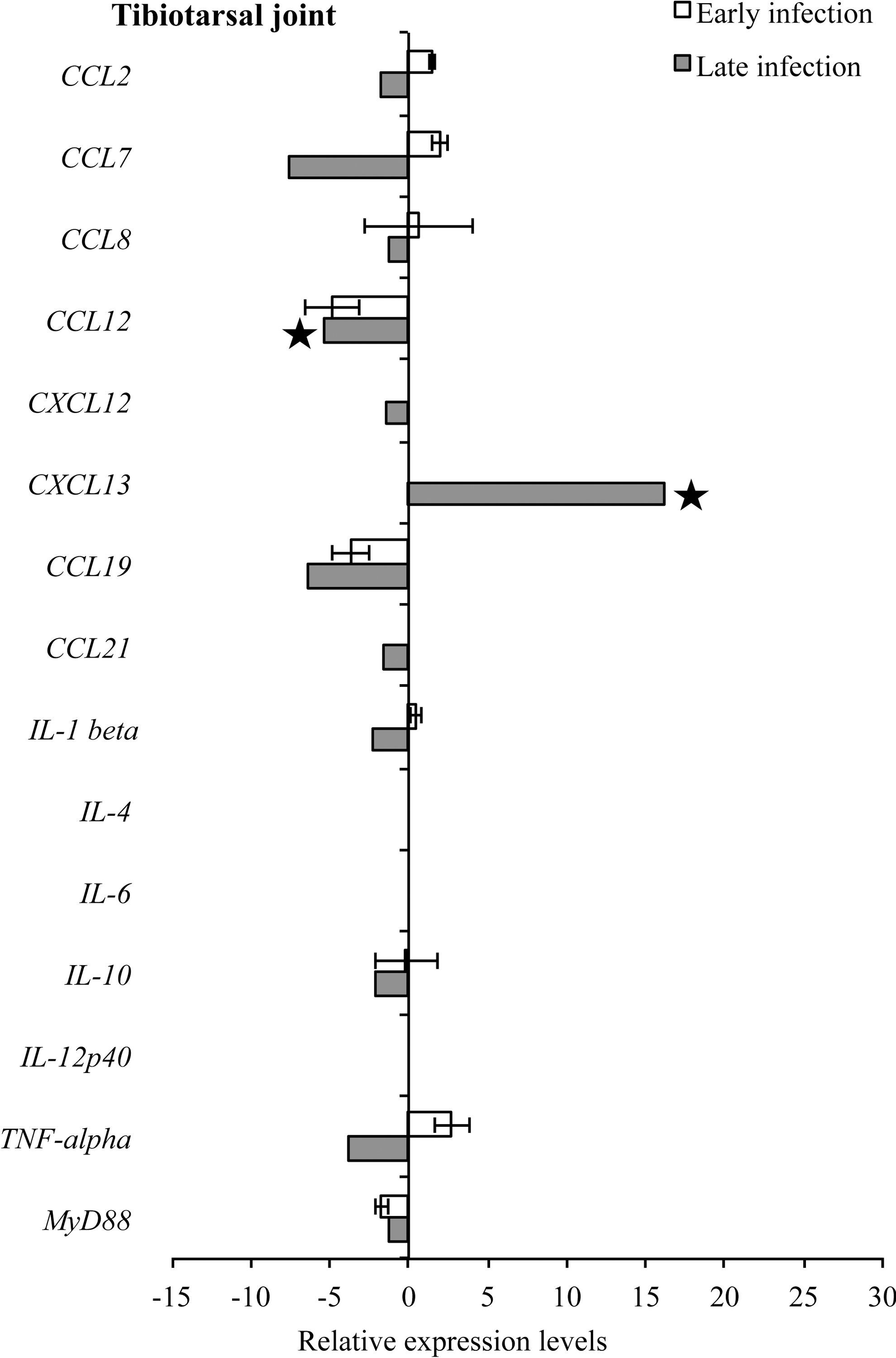
RNA transcription levels (means±standard deviation [SD]) of 19 mouse genes in the tibiotarsal joint of infected C3H mice relative to gene transcription levels in uninfected (sham inoculated) C3H mice during early and late infection. Samples from infected C3H mice were normalized to samples from sham-inoculated mice, expressed in comparison to reference genes (18S rRNA, Actb, B2M, and gapDH), and plotted as fold changes for each gene. Bars to the left of the center line indicate reduced relative expression and bars to the right indicate increased relative expression in infected C3H mice compared to sham-inoculated mice. Zero relative expression indicates no expression in infected C3H mice. Mouse genes not assessed in mice during early infection were CXCL12, CXCL13, CCL21, and IL-21. Transcriptional activity was not detected for chemokines interleukin-2 (IL-2), IL-17a, IL-21, and interferon-γ (IFN-γ in infected C3H and sham-inoculated mice during early and late infection and was not plotted.
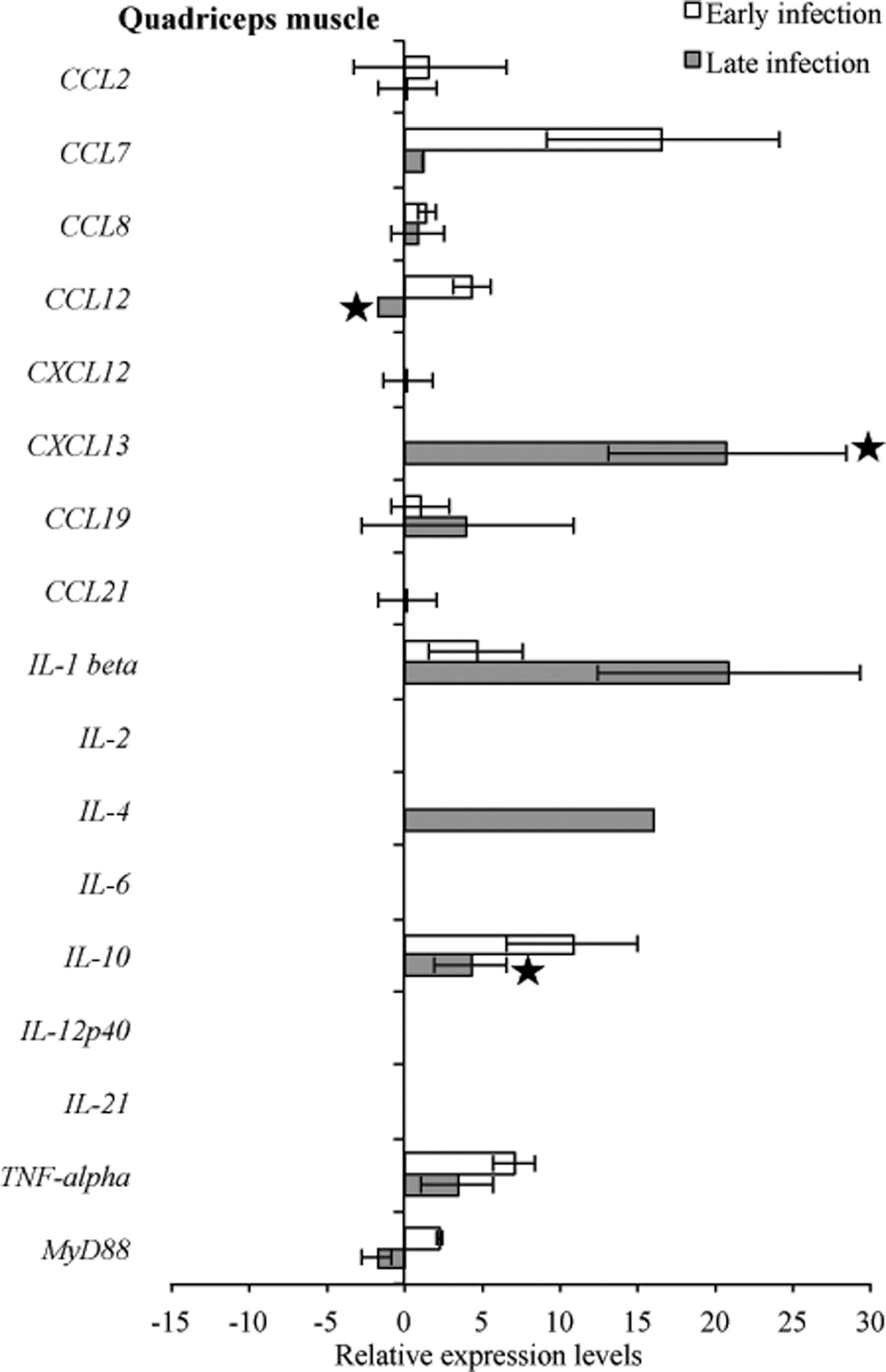
RNA transcription levels (means±standard deviation [SD]) of 19 mouse genes in the quadriceps muscle of infected C3H mice relative to gene transcription levels in uninfected (sham inoculated) C3H mice during early and late infection. Samples from infected C3H mice were normalized to samples from sham inoculated mice, expressed in comparison to reference genes (18S rRNA, Actb, B2M, and gapDH), and plotted as fold changes for each gene. Bars to the left of the center line indicate reduced relative expression and bars to the right indicate increased relative expression in infected C3H mice compared to sham-inoculated mice. Zero relative expression indicates no expression in infected C3Hmice. Mouse genes not assessed in mice during early infection were CXCL12, CXCL13, CCL21, and IL-21. Transcriptional activity was not detected for chemokines interleukin-17a (IL-17a) and interferon-γ (IFN-γ) in infected C3H and sham-inoculated mice during early and late infection and was not plotted.
In summary, LDA revealed variable patterns of host gene expression in different tissues and at different intervals in infected mice.
Discussion
In this study, responses of the pathogen and the host during early and late infection were investigated, comparing relative gene expressions in selected tissues of immunocompetent versus immunodeficient mice, and infected versus uninfected immunocompetent mice. This was performed with LDA assays that included genes of B. burgdorferi cN40 known to be involved in attachment, cell envelope, metabolism, complement regulation, cellular processes, and replication, as well as selected host innate and acquired immune response genes. LDA proved to be sensitive and reproducible and revealed complex patterns of relative response among treatment groups and tissues. Therefore, LDA should be useful for more incisive analysis of B. burgdorferi infection in both the vector and the host.
To study the transcriptional activity of a gene's mRNA, it is appropriate to choose a valid internal control for monitoring intersample variation. Genes that are transcribed with the target genes, but are not regulated by the experimental design, can be used as reference genes (Bustin 2000, Huggett et al. 2005, Gilsbach et al. 2006). For the identification of a suitable reference gene, comparison of transcriptional activity among six selected B. burgdorferi genes (16S rRNA, flaB, gapDH, p13, p66, dbpA) was evaluated. Analysis revealed that transcription of most of these possible reference genes is highly variable. In particular, some of the potential reference genes had tissue-specific patterns. The most stable reference gene selected was 16S rRNA, which appeared to be constantly transcribed at similar range among different tissue types and at the different phases of infection. In several studies, flaB was used as a reference gene for relative quantification, as in nonhuman primates necropsied 2–3 months postinjection (Miller et al. 2005, Miller and Stevenson 2006), in ticks (Gilmore et al. 2001, Koci et al. 2006), and in culture (Hyde et al. 2007). This gene has generally been regarded to be constitutively expressed, but results suggest otherwise. Using RT-qPCR, we reported previously that flaB mRNA transcription was not detected in all tissues and not in all samples, especially those collected during late infection (Hodzic et al. 2003). This study supports our previous finding that flaB is not always constitutively expressed, especially during infection, suggesting that flaB is not an ideal reference gene for studying B. burgdorferi during infection.
Although the 16S rRNA gene was determined to be the most constantly transcribed gene for use as a reference gene, a problem in transcription of ribosomal genes in prokaryotes that are used as reference genes can arise on the mRNA before translation is terminated, causing a ribosome to stall. This can occur when a mutation in the DNA changes a termination codon that specifies an amino acid, or when transcription terminates prematurely. In these cases, the ribosome reaches the end of the mRNA without encountering a termination codon and stalls, still attached to mRNA (Pierce 2008). Vandecasteele et al. (2001) have reported that expression of the 16S rRNA gene in Staphylococcus aureus decreased significantly more rapidly and earlier than the expression of other reference genes. Ideally, expression of a good reference gene should not be altered by the experimental conditions, but many studies have reported that not a single gene is expressed invariantly. The most stable B. burgdorferi reference gene identified for this study was 16S rRNA, with high transcriptional activity and invariant of the condition studied.
LDA is a technological approach capable of measuring transcriptional activity of many genes in a single sample as in microarray, while featuring the sensitivity and quantification attributes to real-time qPCR. In recent years, LDA has been used to study complex gene expression changes under various environmental conditions in a host (Goulter et al. 2006, Steg et al. 2006, Cai et al. 2007). One of the aims of this study was to assess the LDA approach for study of Lyme borreliosis, and the ability of LDA to detect and measure transcriptional activity of 43 B. burgdorferi genes simultaneously in single samples, as well as 19 host genes. Hence, LDA is suitable for examining changes in gene expression that are involved in pathogenesis and persistence, as well as in response to changing environmental and physiological conditions and pharmacological stimuli (Goulter et al. 2006, Steg et al. 2006, Lu et al. 2008, Sanchez-Espiridion et al. 2009). Searching available literature, no publications were found that described the use of LDA to study B. burgdorferi gene expression.
The amounts of B. burgdorferi RNA and cDNA available from collected mouse tissues are limited, especially in immunocompetent mice and during late infection, which restricts the number of analyzable genes. To overcome this problem, a preamplification technique was used to enhance the sensitivity and fidelity of the RT-qPCR, especially for low-abundance target genes, which increases the fidelity of target genes that can be analyzed. Identical primers were used for both the preamplification reaction and the succeeding RT-qPCR. The preamplification approach for gene transcription gave a high yield of cDNA, an increase in sensitivity, low interassay variation, and consistent results. This confirms that preamplification is a suitable tool for investigating gene expression analyses with the limited RNA/cDNA amounts derived from mice with persistent infection and fewer spirochete burdens. Narasimhan et al. (2003) have used a preamplification approach to study B. burgdorferi gene expression in the central nervous system and heart of nonhuman primates. The authors used a different methodology (membrane arrays) than that which was employed here, which makes comparison of results difficult. Ciotti et al. (2009) have reported that preamplification is a powerful and reproducible approach that can be performed even using poorer RNA quality samples.
The present study focused on a variety of genes located throughout the B. burgdorferi genome that encode proteins associated with a variety of functions, during early (3 weeks) and late (2 months) infection in immunocompetent and immunodeficient mice. We used the well-characterized C3H and C3H-scid mouse models, in which inflammation is consistently present in the heart base, but minimal in the ventricular muscle at 3 weeks of infection; inflammation subsides at 2 months of infection (Armstrong et al. 1992). Thus, the model allowed testing the same tissue types during the inflammatory and resolved (persistent) phases of infection. Development of arthritis, however, can be variable, depending upon mouse strain, age, and infectious dose (Barthold et al. 2010). In the current study, arthritis was minimal in C3H mice and was found in only one mouse at the 3-week interval.
Host responses may play roles in the pathogenesis of Borrelia infection, such as inducing expression of lipoproteins on the cell surface, which facilitates dissemination and inflammation. Expression of surface-exposed lipoproteins results in cytokine/chemokine activation that may induce host inflammatory reactions at the pathogen's predilection sites (skin, heart, joint, central nervous system), while their downregulation permits the survival of spirochetes for prolonged periods of time despite the host immune responses (Norris et al. 2010). Comparative analysis of B. burgdorferi gene transcription revealed higher activity in tissues of immunodeficient compared to immunocompetent mice at both early and late infection in all tissues, and for all genes except acrB (not transcribed at either time point) and crasp1 (not transcribed during late infection). These observations support previous findings that transcriptional activity of B. burgdorferi genes have been influenced by the host immune system as a strategy of immune evasion (Liang et al. 2002a, Brooks et al. 2003).
Notably, transcription of two genes in particular, ospA and ospC, in tissues of mice in the current study validated the LDA approach. OspA is expressed by spirochetes within the midgut of ticks, and ospA is downregulated during mammalian infection. One signal for downregulation of ospA in the tick is a natural antibody that is acquired during the blood meal (Belperron and Bockenstedt 2001). In contrast, ospA is transcribed in immunodeficient C3H-scid mice, and it has been found that passive transfer of normal mouse serum in infected C3H-scid mice results in ospA downregulation (Hodzic et al. 2005). OspA also tends to be expressed in the context of inflammation (Crowley and Huber 2003). These patterns of ospA transcription in infected C3H-scid mice were observed in the current study. OspC has been shown to be essential to establishing initial infection, but is vulnerable to a specific antibody, and therefore is downregulated as infection proceeds in immunocompetent hosts (Liang et al. 2004b, Seemanapalli et al. 2010). As expected, ospC was transcribed in C3H-scid mice, but was downregulated in immunocompetent C3H mice as infection progressed. These known patterns of transcription serve as validation of the LDA approach.
Differential expression of B. burgdorferi genes was assessed between genes located on the chromosome versus plasmid. No differences were noted during early infection, whereas during late infection most of the upregulated genes that were identified on plasmids encode proteins involved in complement regulation, and those identified on the chromosome encode proteins involved in attachment, cell envelope, cell processes, and DNA metabolism. Brooks et al. (2003) have reported that almost all of the upregulated genes of B. burgdorferi cultured under different conditions were chromosomally located, and the vast majority of downregulated genes were encoded on plasmids. Revel et al. (2002) reported similar results, in which cultured as well as host-adapted spirochetes in dialysis membrane chambers implanted in rats were used. Liang et al. (2002c) have found that during early infection five of six of the assessed lipoprotein genes were transcribed, while at later infection four of five of those transcribed during early infection were downregulated. Of the downregulated genes, two of three were plasmid-encoded, and one of three was located on a chromosome. A microarray technique has been used to study differential expression of borrelial genes cultivated in different environments (Revel et al. 2002) and exposed to different temperatures (Ojaimi et al. 2003) or to mammalian host factors (Brooks et al. 2003, Tokarz et al. 2004). Our data are difficult to compare with those cited above because they used a different technique, in vitro-grown spirochetes, and different tissue samples and different stages of infection. Nevertheless, LDA offers the advantage of quantitatively assessing an array of genes simultaneously.
Many studies have shown that cytokines play an important role in the pathogenesis of Lyme borreliosis in the initial phase of infection (Miller et al. 2008, Rupprecht et al. 2009, Bachmann et al. 2010), while at late infection expression of some, especially Th1-associated cytokines, waned (Sjowall et al. 2011). One of the most investigated cytokines is IL-10, which was slightly overexpressed (Benhnia et al. 2005, Lazarus et al. 2008), and has been shown to be a main cytokine to regulate inflammatory response in B. burgdorferi infection (Gautam et al. 2011). Comparative analysis of the experimental data of heart tissue revealed higher levels of relative expression for IL-10 during both early and late infection. In the tibiotarsal joint, expression of IL-10 during early and late infection was unchanged when compared to sham-infected control mice, whereas in the quadriceps muscle one out of five samples had higher detectable levels of this cytokine. Lazarus et al. (2006) reported that clearance of B. burgdorferi by innate immunity is more efficient in the absence of IL-10, which could explain our finding of IL-10 downregulation and lack of inflammation in the tibiotarsal joint.
Major proinflammatory cytokines that are involved in induction of inflammatory responses include IFN-γ and TNF-α, which were upregulated and correlated with a greater severity of Lyme disease (Christopherson et al. 2003, Dame et al. 2007). The synergistic effect of B. burgdorferi and IFN-γinduces upregulated expression of numerous genes, of which seven encode chemokines (CCL7, CCL8, CXCL2, CX3CL1, CXCL9, CXCL10, CXCL11) that attract T lymphocytes and neutrophils (Dame et al. 2007). In our study, we demonstrated a significant increase of IFN-γ in heart tissue during early and late infection, along with CCL7 and CCL8, whereas TNF-α was upregulated in heart tissues, except in heart base samples, during late infection.
Comparative analysis of the transcriptional activity of B. burgdorferi and cytokines revealed that increased flaB mRNA during early infection was followed by an increase of CCL7, CCL8, IL10, and TNF-α in all assessed tissue types. Also, during early infection in all tissue tested, most complement regulation genes tested were downregulated. In ventricular muscle during both early and late infection, IL-4 was downregulated as well as B. burgdorferi genes involved in cellular processes (acrB, relA/spot) and cell envelope (bmpC, bmpD, ospC, eppA).
The main aim of this study was to assess the feasibility of LDA to study multiple gene expression of B. burgdorferi during infection and host responses to infection. This work shows complex expression of B. burgdorferi genes that correspond to expression of certain host cytokines and chemokines, of which immunoregulatory effects seem to be optimized for bacterial persistence in the host. These results will pave the way for a more comprehensive study of multiple genes in selected tissues under different conditions and treatments during different stages of infection in this important vector-borne disease.
Footnotes
Acknowledgments
This work was supported by PHS grant R01 AI026815 from the National Institute of Allergy and Infectious Diseases. The technical assistance of Kimberly Olsen, Edlin Escobar, Samantha Mapes, and Alicja Omanska is gratefully acknowledged.
Author Disclosure Statement
The authors have no conflicts of interest in connection with the submitted manuscript.