
Abstract
Introduction:
Arbovirus screening in invertebrate vectors is an important component of the vector-borne disease surveillance programs. Turkey has been shown to harbor medically important mosquito-borne arboviruses such as West Nile Virus (WNV). However, limited information about infections in vectors are currently available. This study was performed to provide preliminary data from Eastern Thrace region, Turkey, where no arbovirus vector surveillance has previously been performed.
Materials and Methods:
Mosquito sampling was undertaken at 23 sites in Edirne province during July, 2012. All specimens were identified morphologically, and selected individuals were subjected to DNA barcoding via cytochrome c oxidase I (COI) sequencing. Consensus PCR for Flavivirus, Alphavirus, and Phlebovirus genera and WNV-specific nested and real-time reverse transcription PCR were employed for mosquito pool screening and/or confirmation. Viral sequences detected in pools were characterized via sequencing.
Results:
A total of 9261 mosquitoes were captured and distributed into 232 pools from the following species: Ochlerotatus caspius (90.9%), Culex pipiens sensu lato (s.l.) (4.7%), Anopheles pseudopictus (3%), and Anopheles maculipennis s.l. (1.3%). Specimens morphologically classified as Cx. pipiens s.l. were identified as Cx. pipiens pipiens via barcoding. Thirty-seven mosquito pools (15.9%) were positive in pan-flavivirus and WNV-specific assays. Viral sequences in positive pools were characterized as WNV lineage 1 clade 1a and demonstrated 1–4% divergence. No flavivirus sequences other than WNV were detected in the mosquito pools. WNV infection rates in Oc. caspius and Cx. pipiens s.l. pools were 15.6% and 36.3%, respectively. Comparison of current and previously identified WNV sequences from Turkey revealed 94.00–96.34% similarity.
Discussion:
WNV RNA was identified for the first time in Cx. pipiens s.l. and Oc. caspius mosquitoes in Eastern Thrace, Turkey. Our findings indicate the circulation of WNV lineage 1 strains in potential vector species and provide an epidemiological link between WNV activity in mosquitoes and vertebrate infections.
Introduction
Serologic data has shown that Turkey harbors many medically important arboviruses. However, vector surveillance studies targeting arboviruses are scarce, especially those related to mosquitoes and sandflies (Ergunay et al. 2011). Reports of symptomatic WNV infections since 2009 (Ergunay et al. 2011, Ergunay et al. 2012, Kalaycioglu et al. 2012) have demonstrated the potential of mosquito-borne diseases to become significant public health problems requiring the surveillance of potential virus vectors. Turkey's geographical location, climate, and established arthropod populations (Ramsdale et al. 2001) not only provide the potential to amplify arboviruses already in circulation but also suitable habitats for the introduction and emergence of new viruses (Tilston et al. 2008).
To obtain data about the activity of various medically important arboviruses and their vectors, this study was conducted as a pilot cross-sectional surveillance project in Eastern Thrace, Turkey, where no such data have previously been available. This region, directly neighboring Greece (Fig. 1), where WNV emergence has been previously noted (Danis et al. 2011), is also the site where the presence of the Asian tiger mosquito Aedes albopictus (Stegomyia albopicta), the recognized vector of CHIKV transmission in Europe (Bonilauri et al. 2008), has recently been identified (Oter et al., 2013).
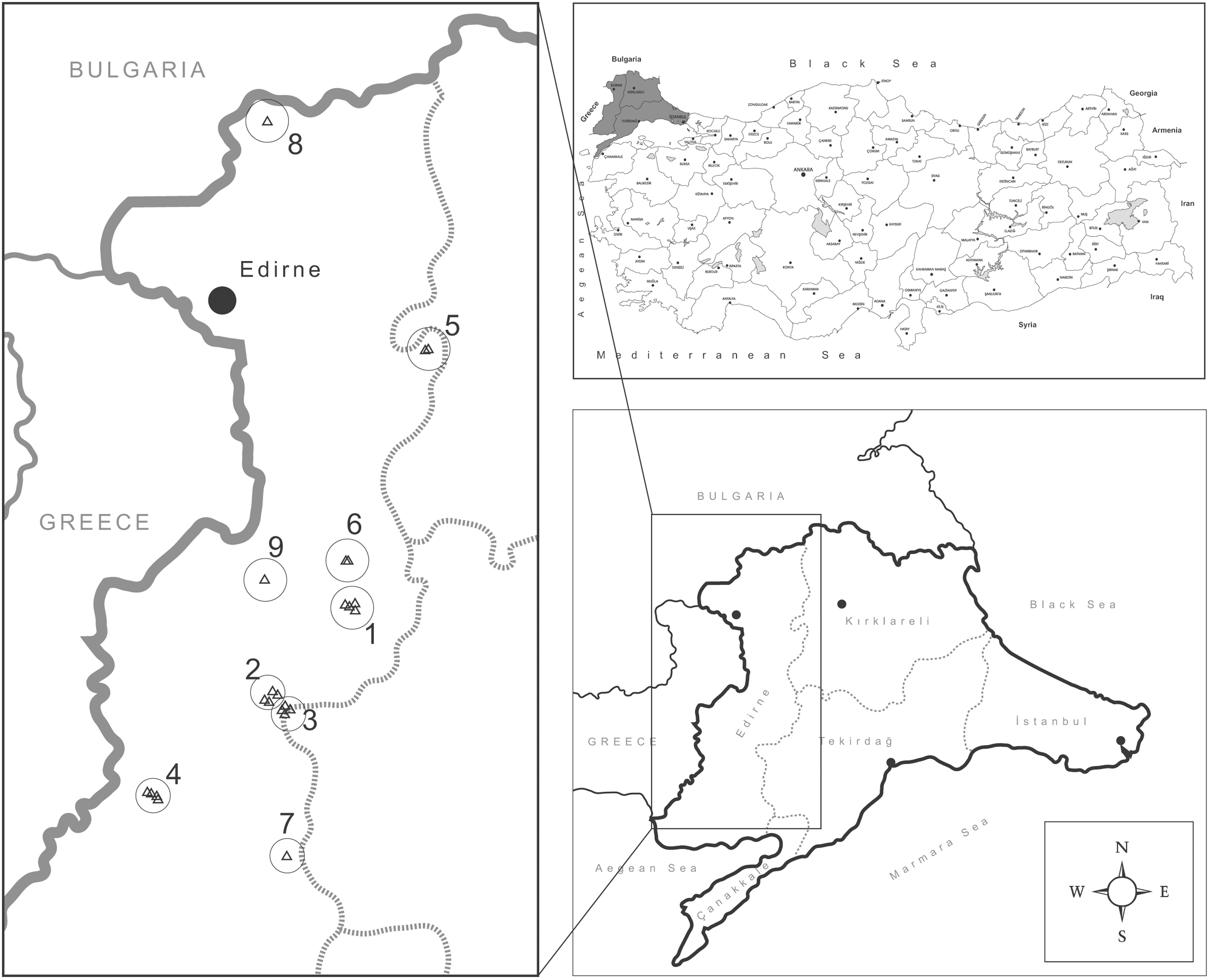
Eastern Thrace, Edirne province, and mosquito sampling locations.
Materials and Methods
Setting and sample collection
The study was performed in the western region of the Turkish Republic, the Eastern Thrace region, with a land area of 23,764 km2 (roughly 3% percent of Turkey's territory) and a population of 1,569,388 in 2011. Eastern Thrace includes five provinces (Edirne, Tekirdag, Kirklareli, Canakkale, and Istanbul), 26 districts, and 678 villages (
Mosquito sampling was performed for 5 consecutive days between July 3 and July 8, 2012, at 23 sampling sites at nine locations (Fig. 1) in urban and suburban environments around villages. CDC Miniature Light Traps (John W. Hock Company, Gainesville, FL) were employed at each site, and aspirators were used for collecting adult mosquitoes from inside and outside houses and barns. Traps were placed 1–2 meters above ground in peridomestic sites and left on site from 18:00 to 06:00 each night. Captured mosquitoes were kept alive, transferred to the laboratory on ice, and identified morphologically to species level using published keys (Darsie and Samanidou-Voyadjoglou 1997, Schaffner et al. 2001). Subsequently, mosquitoes were pooled according to the collection site and species to include 30–40 individuals regardless of sex, and stored at −80°C until processing.
Mosquito pools were homogenized as described previously (Calzolari et al. 2010a) and clarified by centrifugation at 4000 rpm for 4 min. Supernatant from each pool was used for nucleic acid purification using a High Pure Viral Nucleic Acid Kit (Roche Diagnostics, Mannheim, Germany), followed by reverse transcription via random hexamers using a RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific, Tokyo, Japan). All commercial assays were performed according to the manufacturers' instructions.
For confirmation of the morphological identification of the mosquito species, a 658-bp sequence of the cytochrome c oxidase I (COI) gene used for DNA barcoding was amplified as described previously (Folmer et al. 1994) with primers LCO1490 and HCO2198. Two randomly selected mosquito specimens from all species identified at each sampling site were processed and amplified individually. PCR products were purified using a High Pure PCR Product Purification Kit (Roche Diagnostics, Mannheim, Germany) and subjected to cycle sequencing with forward and reverse primers.
Virus detection and characterization in captured mosquitoes
Mosquito pool screening
Three sets of degenerate primers targeting the conserved regions encoding viral polymerases were employed for nested PCR detection of different virus genera in mosquito pools, as previously described (Table 1). In addition, a WNV-specific nested PCR targeting E protein–coding virus genome region was used for screening all mosquito pools (Table 1). Amplifications were performed using Taq DNA polymerase (Thermo Scientific, Tokyo, Japan), in a PTC-200 Thermal Cycler (MJ Research, MA). TOSV ISS.Phl.3 and WNV NY99-4132 isolates, grown on Vero cells (ATCC CCL81), were used as positive controls for WNV-specific, pan-flavi and pan-phlebovirus PCR. Complementary DNAs from CHIKV strain LR2006-OPY1 and tick-borne encephalitis virus (TBEV) strain K23 (kindly provided by N. Litzba and M. Niedrig from Robert Koch Institute, Berlin, Germany) were also used in assay optimization and as positive controls. The amplicons were visualized under ultraviolet light after electrophoresis in 1.5–2% agarose gels, depending on the expected size for each primer set. Extreme care was taken to prevent carry-over contamination.
Wobble bases in oligonucleotides are: I, Inosine; Y, C/T; R, A/G; W, G/C; M, A/C.
FAM, 6-carboxyfluorescein; BHQ-1, Black Hole Quencher 1.
Confirmatory assays in reactive mosquito pools
Samples positive in WNV-specific or pan-flavivirus PCR were further subjected to amplification by in-house WNV-specific one-step real-time RT-PCR for the confirmation of WNV reactivity. A set of primers and probes, designed to amplify lineage 1 and 2 WNV strains, were employed (Table 1). The assay was carried out using QuantiTect Probe RT-PCR kit (Qiagen, Germany) with 4 μL of total RNA from mosquito pools, 0.6 μM of each primer, and 0.2 μM of the TaqMan probe. A Rotor-Gene 6000 instrument (Corbett Research, Australia) was employed using the following steps: Reverse transcription at 50°C for 30 min; initial denaturation at 95°C for 10 min, followed by 45 cycles of denaturation at 95°C for 20 s; and annealing at 57°C for 60 s. Fluorescent emission for each sample was measured after each annealing step. Dilutions of extracted RNA from culture-grown WNV were employed as positive controls in tests.
Sequencing and phylogenetic analysis
All positive samples in screening PCRs were characterized by sequencing. For this purpose, PCR products were cleaned up via a High Pure PCR Product Purification Kit (Roche Diagnostics, Mannheim, Germany), ligated to pJET1.2 vector supplied in a CloneJET PCR Cloning Kit (Thermo Scientific, Tokyo, Japan), and used to transform cells, as directed by the manufacturer. Five to 10 colonies were selected for each amplicon and sequenced using p.JET1.2 forward and reverse primers in an ABI PRISM 310 Genetic Analyzer (Applied Biosystems, CA). The sequences obtained were aligned and analyzed using CLC Main Workbench v5.5 (CLCBio, Aarhus, Denmark), with the Jukes–Cantor substitution rate model with 500 bootstrap replicates. Maximum likelihood trees were generated based on the Unweighted Pair Group Method with Arithmetic Mean (UPGMA) tree.
Results
Distribution of mosquito species
A total of 9261 mosquitoes were captured from 23 sampling sites (Fig. 1) and distributed into 232 pools. Morphological identification of the captured mosquitoes revealed activity of four species: Ochlerotatus caspius (211/232, 90.9%), Culex pipiens sensu lato (s.l.) (11/232, 4.7%), Anopheles pseudopictus (7/232, 3%), and Anopheles maculipennis s.l. (3/232, 1.3%). The morphological identification of Oc. caspius from each site was confirmed, and specimens morphologically identified as Cx. pipiens s.l. were characterized as Cx. pipiens pipiens (Cx. pipiens sensu stricto [s.s.]) via DNA barcoding (data not shown). The number and distribution of mosquito species according to the location and sampling site are presented in Table 2.
Identification and characterization of viral sequences in mosquito pools
All mosquito pools were observed as negative in pan-alphavirus and pan-phlebovirus nested PCRs. A total of 37 pools (37/232, 15.9%) were reactive in pan-flavivirus, and WNV-specific nested and real-time RT-PCRs were made up of 33 Oc. caspius (33/37, 89.2%) and 4 Cx. pipiens s.l. (4/37, 10.8%) pools. No flavivirus other than WNV could be detected. Infection rates in Oc. caspius and Cx. pipiens s.l. pools were 15.6% (33/211) and 36.3% (4/11), respectively. The distribution of positive pools according to location and sampling site is presented in Table 2.
Sequence analysis of cloned WNV-specific amplicons revealed partial sequences of varying lengths (245–251 bp). BLAST and BLASTX searches in GenBank revealed these sequences to be 83–100% identical to various WNV strains. Identical sequences were obtained from all clones analyzed from each amplicon (data not shown). All WNV sequences detected in the mosquito pools were phylogenetically related to WNV lineage 1 clade 1a strains (Fig. 2).

Phylogenic analysis of local West Nile viruses (WNVs) based on partial E protein–coding sequences. Virus strains represented are indicated by name following GenBank accession number. Japanese encephalitis virus JEV-GP78 is included as an outlier. *, sequences characterized in this study; L, localization; S, site, Occ, Oc. caspius, Cxp, Cx. pipiens; Eqn1 and Eqn2, sequences identified in equine infections from Central Anatolia in 2011.
Maximum composite likelihood analyses demonstrated 91–97% similarity between sequences detected in the mosquito pools and various WNV strains of different origins (NY99-AF196835, HUN03-DQ118127, and EGY101-AF260968) (Table 3). Identical sequences were obtained in pools originating from the same sampling site in all locations (data not shown). However, 1–4% variation rates were observed in WNV-positive Oc. caspius or Cx. pipiens s.l. pools between various locations and sampling sites (Table 3), with the greatest WNV sequence diversity (96.4% similarity) being observed between infected Cx. pipiens s.l. and Oc. caspius pools originating from different locations (Table 3).
L, localization; S, site, Occ, Oc. caspius, Cxp, Cx. pipiens; Eqn1 and Eqn2, sequences identified in equine infections from Central Anatolia in 2011.
Comparison of WNV sequences from our mosquito pools with lineage 1 WNV sequences identified in Central Anatolia, Turkey, was also performed. The latter include two sequences (GenBank accession nos. JN828805 and JN828806; Fig. 3) recovered during an equine outbreak of fever with neurological symptoms in Eskisehir province (39°24′N 31°02′E) in 2011 (Ozkul et al. 2012, in press). Here, a relatively higher sequence divergence, with 94.00–96.34% similarity between WNV strains originating from Central Anatolia and Eastern Thrace was noted in the maximum composite likelihood analyses (Table 3). Sequences from Eastern Thrace and Central Anatolia were observed to form a distinct cluster within Clade 1a WNV strains (Fig. 2).

Multiple alignment of nucleotide sequences of E protein–coding region of local West Nile viruses (WNVs) detected in Eastern Thrace and Central Anatolia, Turkey, with lineage 1 viruses of various origins (NY99, HUN01, and EGY101). Dots indicate coincident nucleotide. (See Fig. 2 legend for abbreviations.)
Discussion
Vector surveillance studies are of paramount importance to understand the dynamics of autochthonous arbovirus circulation and to predict potential introduction of nonautochthonous viruses via mosquitoes in affected regions. This study was carried out to reveal virus circulation in mosquito populations in Eastern Thrace, Turkey. To our knowledge, this is the first study performed in this region and the most comprehensive study undertaken in Turkey for mosquito screening for arboviruses of various genera, including alphaviruses (Ergunay et al. 2011).
During July, 2012, we captured 9261 mosquitoes from 23 sampling sites at 9 locations, distributed mainly around Edirne province, close to Turkey's borders with Bulgaria and Greece (Fig. 1). Morphological identification of all individuals revealed Oc. caspius to be the most abundant mosquito species (90.9%), followed by Cx. pipiens s.l. (4.7%), An. pseudopictus (3%), and An. maculipennis s.l. (1.3%). DNA barcoding of selected specimens confirmed the morphological identification of Oc. caspius, whereas Cx. pipiens s.l. specimens were characterized as Cx. pipiens s.s., belonging to the Cx. pipiens complex with Cx. pipiens modestus and Cx. pipiens quinquefasciatus. Although the mosquito species of Eastern Thrace have not been thoroughly investigated, activity of all identified species in this study has previously been reported from various other regions in Anatolia (Ramsdale et al. 2001). For example, during an active surveillance study in 2011, eggs of Ae. albopictus were detected in two separate localities in the region, including the Turkish–Greek border at Ipsala (40°55′N and 26°22′E) and in the vicinity of the Kesan district (40°51′N 26°38′E), 20–100 km away from the current sampling sites (Oter et al., 2013). No Ae. albopictus specimens were identified in this study. The clear preponderance of Oc. caspius over other mosquito species across a relatively large sampling area is another notable finding of this study (90.9% vs. 9.1%). The main limitation of the study is that only a single field survey was performed throughout the entire season, meaning that the species frequencies and distributions represent cross-sectional data. Therefore, it remains to be determined whether mosquito species identified in this study reflect the pattern of species circulation in Eastern Thrace.
For arboviral surveillance, the pooled mosquitoes were investigated for the presence of viral RNAs from Alphavirus, Flavivirus, and Phlebovirus genera via nested consensus PCRs using primers that target conserved regions of the viral polymerase enzymes (Table 1). Mosquito pools were also screened via a WNV-specific nested PCR based on the E protein–coding genome region (Table 1). No alphavirus or phlebovirus sequence was detected. In 15.9% of the screened pools, WNV-specific nested PCR displayed positive results that were further confirmed via real-time RT-PCR. The positive pools consisted of two mosquito species, Oc. caspius (89.2%) and Cx. pipiens s.l. (10.8%). Sequencing of the amplicons demonstrated that all strains belong to lineage 1 clade 1a viruses (Fig. 2). Limited sequence divergences were noted between mosquito species from identical or different sampling locations (Fig. 3). Comparison of WNV sequences identified in mosquitoes and in equine infections from Central Anatolia in 2011 suggests Oc. caspius and/or Cx. pipiens s.l. species are the mosquito vectors probably responsible for virus transmission. This is the first detection of WNV nucleic acids in mosquitoes, establishing an epidemiological connection between probable vectors and vertebrate infections in Turkey, which was not possible in previous studies. For example, WNV antigens or nucleic acids could not be detected in 6457 mosquito specimens of Cx. pipiens s.l., Oc. caspius, and Aedes species captured in Sanliurfa province (southeastern Anatolia), although human seroreactivity was observed (Ozer et al. 2007). Similarly, no viral RNA could be found in samples from various wild avian species from the Kizilirmak delta (Albayrak and Ozan 2010). Finally, 4698 mosquitoes (99.6% of which were Cx. pipiens s.l.) captured across western Anatolian provinces associated with a recent WNV outbreak (Kalaycioglu et al. 2012) proved negative for WNV RNA (Y. Ozbel. Presentation at the West Nile virus Symposium, September 20–21th, 2012 Ankara, Turkey). For a comprehensive analysis of WNV circulation dynamics in Eastern Thrace, WNV surveillance needs to be performed throughout the season to reveal periods of peak activity, which was not possible in this study.
Among the mosquito species distributed throughout Europe, Cx. pipiens s.s., Cx. theileri, Cx. modestus s.s., Cx. univittatus, Oc. caspius, and An. maculipennis s.l. species are considered to be competent WNV vectors and appear to play an important role in virus transmission (Calistri et al. 2010a). In Italy, WNV was detected in Cx. pipiens and Oc. caspius pools located in around the region of an human outbreak (Monaco et al. 2010), and subsequent studies revealed the expansion of areas where infected mosquitoes could be found (Calistri et al. 2010b, Calzolari et al. 2010b). In France, Cx. modestus and Oc. caspius are considered the main vectors in wetland areas and Cx. pipiens in urban and periurban areas (Balenghien et al. 2008). WNV lineage 1 viruses were identified in Cx. perexiguus mosquitoes in Spain, suggesting this species as the vector for WNV in Spain (Vasquez et al. 2011), whereas in neighboring Portugal, the activity of Cx. pipiens, An. maculipennis s.l., and Cx. theileri were associated with human WNV cases (Almeida et al. 2008). In Greece, where WNV emergence coincided with Turkey, infected Cx. pipiens s.l. mosquitoes were detected at one outbreak location (Papa et al. 2011). In the Volgograd region of Russia, Cx. pipiens s.s. was implicated in WNV transmission in urban areas and Cx. modestus s.s. in periurban areas (Fyodorova et al. 2006). Cx. perexiguus, Cx. pipiens, and Oc. caspius species were shown to be the WNV vectors in Israel, without any association of seasonal abundance and infection (Orshan et al. 2008).
The present study adds to this knowledge by identifying WNV sequences in 36.3% of the Cx. pipiens s.l. pools during June–August, 2012, in Eastern Thrace, Turkey. We also observed an abundance of Oc. caspius species with a WNV infection rate of 15.6%. Although in vitro studies indicate that Oc. caspius is an inefficient WNV vector (Balenghien et al. 2008), WNV sequences have consistently been detected in Oc. caspius mosquitoes in various countries (Balenghien et al. 2008, Monaco et al. 2010) and have been suggested as a probable vector (Orshan et al. 2008). Thus, the present study's findings indicate that the impact of Oc. caspius in WNV epidemiology in Turkey should be addressed in prospective studies. Interestingly, while lineage 2 WNV activity has been reported in human cases, as well as in infected Cx. pipiens s.l. mosquitoes from neighboring Greece (Papa et al. 2011), we have detected lineage 1 strains in Eastern Thrace, on the eastern border of the Balkan peninsula. It is known that WNV dispersion within a region is directly influenced by the origin of migrating bird species and seasonal aggregations in similar or distinct habitats (Jourdain et al. 2007). The circulation of various WNV lineages can also be observed within a geographical area (Magurano et al. 2012). More information is needed to fully understand WNV dynamics and dispersion in Eastern Thrace region.
This study's findings indicate the circulation of lineage 1 viruses in Eastern Thrace as well as in Central Anatolia. However, these analyses were based on partial sequences, so larger segments are required for a more comprehensive phylogenetic evaluation. Virus isolation and whole genome sequencing efforts are underway by our group to reveal the properties of the viruses in circulation.
Conclusions
This study has revealed the circulation of lineage 1 viruses in Eastern Thrace, Turkey, through detection of identical and/or similar WNV sequences in Cx. pipiens and Oc. caspius mosquitoes. For an extensive analysis of the species and viral dispersion in this region, surveillance studies encompassing the full mosquito season are required.
Footnotes
Acknowledgments
This work was supported by a grant from the Scientific and Technological Research Council of Turkey (TUBITAK, SBAG 110S404). The authors are grateful to Irfan Atmaca and Salim Calis for technical assistance and N. Emin Guven for graphics. Preliminary findings of the study were accepted as a poster presentation at the 23rd European Congress of Clinical Microbiology and Infectious Diseases (ECCMID), to be held in Berlin, Germany between 27-30th of April, 2013.
Author Disclosure Statement
All authors declare that they have no competing financial interests.