
Abstract
We hereby propose a novel sensitive, specific, and cost-efficient method to detect Ehrlichia canis and Anaplasma platys DNA from canine whole blood samples by multiplex PCR. Blood samples from hemoparasited dogs attending the Veterinary Hospital at the Universidade Federal Rural da Amazônia–UFRA, Belém, Brazil, were collected in tubes containing EDTA. Amplification of E. canis and A. platys 16S rDNA by nested (n) PCR was successfully achieved by using primers specific to the Anaplasmataceae in the first round of PCR, followed by a second round of PCR using E. canis–specific primers in conjunction with A. platys–specific primers. The amplicons obtained were cloned and sequenced, yielding sequences of 478 and 473 bp (including primers) pertaining to regions of the 16S rDNA of E. canis and A. platys, respectively. The protocol we here propose may help to measure the prevalence of canine monocytic ehrlichiosis (CME) and canine cyclic thrompocytopenia, not only in northern Brazil, where there is no data available, but also elsewhere.
Introduction
E
CCT is characterized by cyclical parasitemia in platelets, followed by generalized thrombocytopenia and lymphadenomegaly (Baker et al. 1987). CCT can vary clinically from mild to severe (Dumler et al. 1995, Ferreira et al. 2008). In its chronic phase, a few days after infection, there is a sudden decrease in the number of platelets and the etiologic agent, which configures sporadic parasitemias (Harvey et al. 1978, Chang et al. 1996). In its chronic phase, platelet counts approach reference values in around 4 days (Harrus et al. 1997, Inokuma et al. 2000). Because of the cyclical nature of CCT, most dogs infected with this form of the disease remain asymptomatic, which renders CCT a relatively benign disease. When present, clinical signs presented by infected dogs include emesis, diarrhea, fever, depression, and anorexia, and laboratory findings are, in addition to cyclic thrombocytopenia, usually nonregenerative normocytic normochromic anemia, discrete leucopenia, hypoalbuminemia, and hyperglobulinemia (Rikihisa 1991, Chang and Pan 1996, Harvey 2006).
Although these clinical signs may be similar to those presented by other diseases (Rikihisa 2000), differential diagnosis is highly relevant not only from a clinical standpoint but also from an epidemiological one. Traditionally, diagnosis of infection by E. canis and A. platys is usually achieved through observation of clinical signs, by serological methods, and by direct detection in blood smears (Waner et al. 2001). However, these diagnostic criteria are often ambiguous and may fail to identify the pathogen species involved. Additionally, one must pay careful attention to the detection of basophilic inclusions in smears of blood platelets in whole blood or buffy coat, as intranuclear inclusion bodies of E. canis can be observed in platelets (Souza et al. 2004). Also, one should take into account that the infected dog platelets have a cyclical nature, the observation of basophilic inclusions in smears of blood platelets in some phases of the disease not being possible (French and Harvey 1983, Chang et al. 1996).
In this context, more precise diagnostic methods, such as molecular diagnosis have been developed (Chang and Pan 1996, Murphy et al. 1998), which have gained great importance due to the growing interest in the study of these bacteria as causative agents of infections in humans (Tami and Tami-Maury 2004). Because Rhipicephalus sanguineus ticks are considered the primary vector of both pathogens (Groves et al. 1975, Greene and Harvey 1990), coinfection of E. canis and A. platys may be common in some regions, although to the best of our knowledge there is currently no molecular protocol to specifically detect these etiological agents in a single test. Thus, in this study we propose a sensitive, specific, and cost safe method to detect E. canis and A. platys with multiplex PCR.
Methods
Blood samples were collected in EDTA tubes from dogs attending the Veterinary Hospital of the Universidade Federal Rural da Amazônia–UFRA. Total DNA was isolated from 250 μL of whole blood with an AxyPrep™ Blood Genomic DNA Miniprep Kit (Axygen), according to the manufacturer's instructions. For this study, we selected samples positive for E. canis only (n=10), A. platys only (n=10), and E. canis/A. platys coinfection (n=10). The positivity of the controls were confirmed based on the comparison of the nucleotide sequences of the fragments amplified with the Ecan/HE3 and ApysF/ApysR primers with sequences obtained from GenBank under accession numbers EF195134 and JQ894779, respectively. Individual detection of E. canis and A. platys was performed according Murphy et al. (1998) and Chang and Pan (1996), respectively.
Amplification of E. canis and A. platys 16S rDNA regions in a multiplex PCR was achieved using ECC and ECB primers (Dawson et al. 1994, 1996), which are specific to Anaplasmataceae taxa, in the first round of PCR followed by a second round of PCR using E. canis-specific primers (Ecan [Murphy et al. 1998] and HE3 [Anderson et al. 1992]) in conjunction with A. platys–specific primers (ApysF and ApysR; this study). ApysF (5′-GTCGAACGGATTTTTGTCGT-3′) and ApysR (5′-TAGATCACCGCCTTGGTAGG-3′) were designed using the Primer3plus software (
The first round of amplification was carried out in 25-μL reactions with 10–20 ng of the DNA template, 2 mM of MgCl2, 2.5 mM each of deoxyribonucleotide triphosphates (dNTPs), 10 mM Tris-HCl, 50 mM KCl, 2.5 μM of each primer (ECC and ECB), and 1.25 U Taq DNA polymerase (Invitrogen). The amplification reaction consisted of 30 cycles of 1 min at 94°C, 2 min at 65°C, and 1 min at 72°C, preceded by 3 min at 94°C and followed by 5 min at 72°C. The second round of amplification was also carried out in 25-μL reactions with 1 μL of the first-round PCR product, 1.5 mM of MgCl2, 5 mM each of dNTPs, 10 mM Tris-HCl, 50 mM KCl, 2.5 μM of each primer (Ecan, HE3, ApysF, and ApysR), and 1.25 U Taq DNA polymerase (Invitrogen). The amplification reaction consisted of 10 cycles of 1 min at 94°C, 1 min at 62°C, and 1 min at 72°C, followed by 35 cycles of 1 min at 94°C, 1 min at 60°C, and 1 min at 72°C. As in the first round of PCR, the 45 cycles of the second PCR were preceded by 3 min at 94°C and followed by 5 min at 72°C. All amplicons were electrophoresed in 1.5% agarose gel and visualized by GelRed™ Nucleic Acid Stain (Biotium) under ultraviolet (UV) light.
To evaluate the specificity of the protocol proposed here, amplicons of the first-round PCR of samples individually infected by E. canis and A. platys, in addition to amplicons of the second-round PCR of coinfected samples, were excised from the agarose gel, and then purified with a GFX PCR DNA and Gel Band Purification Kit (GE Healthcare). An aliquot of each purified product was ligated into a pGEM-T vector (Promega) overnight and then desalted before electroporation into Escherichia coli TOP10 (Invitrogen Life Technologies). The inserted DNA of white clones was obtained by PCR directly from the colonies using M13F/M13R primers and sequenced automatically in a 3130 Genetic Analyzer (Applied Biosystems), according to the manufacturer's specifications. BioEdit software (Hall 1999) was used to align forward and reverse sequences.
Results
Sequencing of the first-round PCR amplicons of E. canis and A. platys yielded sequences, including primers, that were 478 bp long (GenBank accession number KC109445) and 473 bp long (GenBank accession number KC109446), respectively. Alignment of all sequences resulted in one haplotype to E. canis and one to A. platys. Additionally, their pairwise comparison with the sequences retrieved from GenBank returned one transition (A↔G) to E. canis sequences and no polymorphic sites when compared to A. platys sequences. Sequences obtained from the second-round PCR confirmed the amplification of regions of the 16S rDNA of E. canis and A. platys, yielding sequences, including primers, of 389 and 212 bp, respectively.
Even though there is advantage in using a single reaction to detect E. canis and A. platys using the ECC/ECB primers (first reaction), the amplicons generated by either of these two species differ in size by only 5 nucleotides, which makes resolution of the bands impossible by agarose gel electrophoresis. Thus, presence of either or both of these pathogens in a sample yields a single band of ≈500 bp, as shown in Figure 1 (lanes 2 and 3).
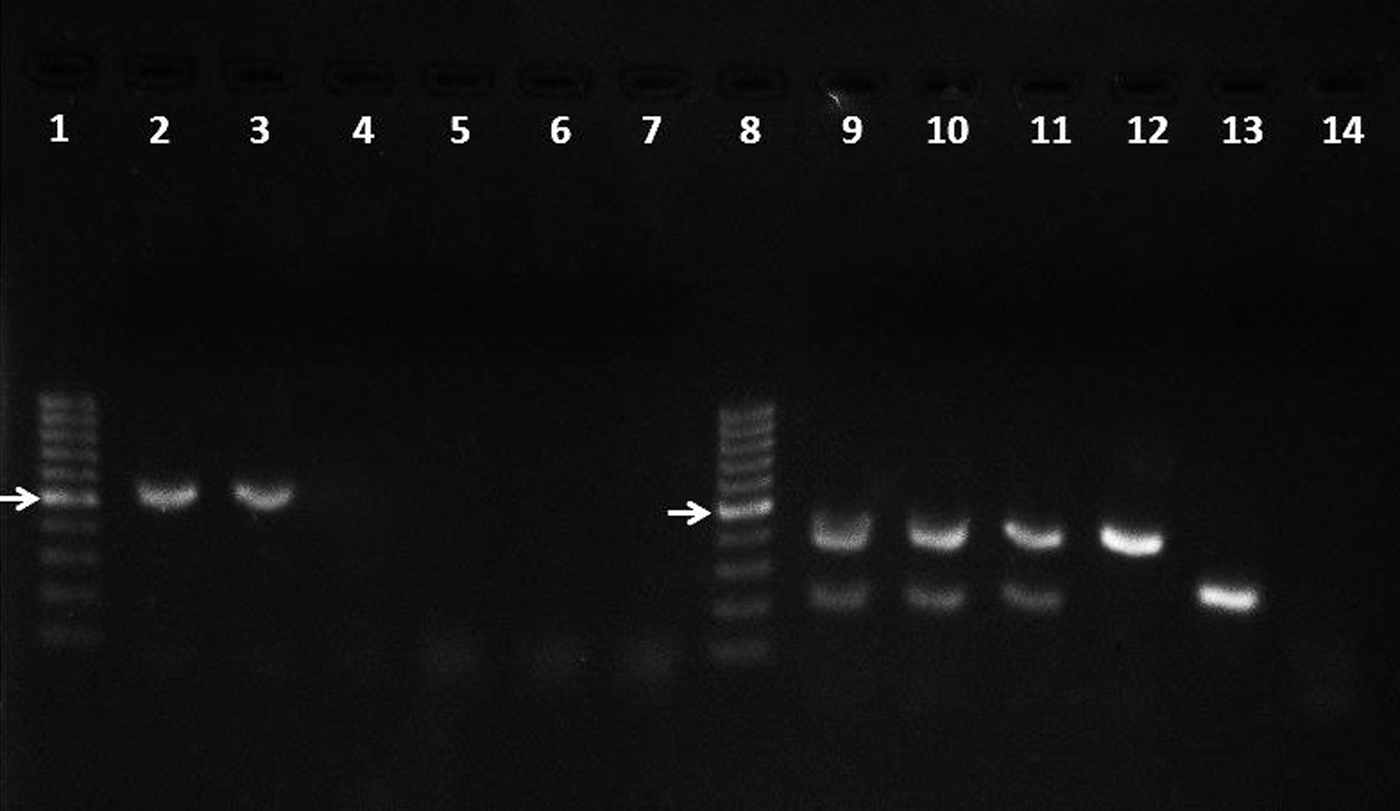
Detection by nested PCR of E. canis and A. platys DNA in the blood specimens of dogs naturally infected. Lanes 2–7 correspond to the first round of PCR, whereas lanes 9–14 correspond to the second round of PCR. Lanes 1 and 8, 100-bp ladder (Invitrogen); lanes 2 and 9, positive control (DNA template obtained from a dog hemoparasited by E. canis and A. platys); lanes 3, 4, 10, and 11, E. canis/A. platys (coinfection) samples; lanes 5 and 12, E. canis only; lanes 6 and 13, A. platys only; lanes 7 and 14, negative control (no DNA template). The arrow corresponds to 500-bp fragment.
In the second reaction, which uses primers Ecan/HE3 and ApysF/ApysR that are, respectively, specific to E. canis and A. platys, a single 389-bp amplicon is observed for E. canis, whereas a 212-bp amplicon is yielded for A. platys (Fig. 1, lanes 12 and 13). In case of co-infection, the reaction yields two resolvable amplicons (Fig. 1, lanes 9–11). It is interesting to note that some samples may result in no amplification in the first reaction (Fig. 1, lanes 4, 5, and 6); however, a clear sign of infection is observed after the second reaction (Fig. 1, lanes 11, 12, and 13), this being observed for 24 of the 30 samples analyzed.
Discussion
Although not the main objective of this study, one of its most important findings is the first reported occurrence of E. canis and A. platys infecting dogs in Belém, State of Pará, in the Amazon region in northern Brazil, where the tropical climate should favor the occurrence of the arthropod vectors such as the brown tick R. sanguineus. In Brazil, depending on the size of the population studied, the prevalence of E. canis and A. platys when detected by blood smear ranges from 2.0% (Oliveira et al. 2000) to 16.0% (Soares et al. 2006) and 1.81% (Pichotano et al. 2004) to 50% (Mundim et al. 2008), respectively. On the basis of the nested (n) PCR results, these prevalences range from 7.8% (Carvalho et al. 2008) to 88% (Dagnone et al. 2009) for E. canis and 15.84% (Ferreira et al. 2007) to 55% (Ramos et al. 2009) for A. platys. Even though these data do not necessarily correspond to the same sample populations, they support the advantage of molecular tests for the detection of arthropod-transmitted bacterial agents. In this context, because the 16S rDNA sequences from E. canis and A. platys are highly conserved (Unver et al. 2003), the protocol proposed here may help to quantifying the prevalence of CME and CCT, not only in northern Brazil, where data is currently unavailable, but also elsewhere.
Despite previous optimization of the molecular method by other authors, this is the first description of a molecular protocol to detect E. canis and A. platys in a two-round multiplex PCR, and our approach proved to be more efficient than those based on a single round of standard PCR. In fact, in comparison to other diagnostic methods, several studies have shown the highest efficiency of nPCR for detection of E. canis and A. platys (Chang and Pan 1996, McBride et al. 1996, Martin et al. 2005, Ferreira et al. 2007, Ramos et al. 2009).
Overall, the protocol we propose here is cost effective and especially useful for veterinary hospitals, which still have relied predominantly on direct diagnosis by blood smears and serological methods. Our protocol has the advantage of detecting E. canis and A. platys concomitantly with great sensitivity and specificity. Thus, we suggest the use of this protocol as a routine laboratory test that will help the veterinarian to: (1) Monitor the effectiveness of antibiotic treatment; (2) obtain the diagnosis of ehrlichiosis before it becomes chronic; and (3) prevent postsurgical bleeding. Ultimately, this preventive measure can generate a better quality of life for the animals living in regions with a high prevalence of these etiological agents.
Footnotes
Acknowledgments
The authors are grateful to FAPESPA (ICAAF 012/2012) for financial support and to CAPES for MSc stipends supporting Claudia Rufino and Pablo Moraes.
Author Disclosure Statement
No competing financial interests exist.