
Abstract
To determine the occurrence of feline bartonellosis in Israel, blood samples were collected from 179 stray and 155 domestic cats from 18 cities or villages in central and northcentral Israel. Samples were screened for Bartonella infection by culture isolation and molecular detection using high-resolution melt (HRM) real-time PCR assay targeting the 16S–23S rRNA internal transcribed spacer (ITS). All positive samples were confirmed by two additional HRM real-time PCR assays targeting two fragments of the β-subunit of RNA polymerase (rpoB) and the 16S rRNA genes. The prevalence of Bartonella spp. infection in the general tested population was 25.1% (84/334). A higher prevalence was detected in the stray (30.7%; 55/179) than the domestic cats (18.7%; 29/155). Bartonella henselae, Bartonella clarridgeiae, and Bartonella koehlerae were highly prevalent in both cat populations, however their distribution among the two populations varied significantly (p=0.016). B. clarridgeiae and B. koehlerae were found to be more prevalent in stray than domestic cats, whereas B. henselae was evenly distributed. Co-infection with two or more different Bartonella spp. was determined in 2.1% (7) of the cats. The ITS HRM real-time PCR assay used in this study was shown to have a greater screening power than bacterial isolation, detecting 94.0% (79/84) compared to 35.7% (30/84), respectively, of all positive samples. The high prevalence of these zoonotic Bartonella species, coupled with the overpopulation of stray cats, and increased numbers of domestic cats in the major urban centers in Israel represent a significant threat for the public health in this country.
Introduction
B
In general, the diagnosis of Bartonella infection has been assessed by isolation of the organism in bacterial culture or by amplification of Bartonella-specific DNA fragments using PCR assays, but both have presented some limitations (Clarridge et al. 1995, Breitschwerdt 2008, Guptill 2010). Bartonella spp. isolation by culture is laborious, often insensitive, and requires a long incubation period (up to 6–8 weeks) due to its fastidious nature (Diaz et al. 2012). On the other hand, conventional PCR assays have been extensively applied for the detection of Bartonella DNA sequences (e.g., citrate synthase [gltA], β-subunit of RNA polymerase [rpoB], 16S rRNA genes and the 16S–23S rRNA internal transcribed spacer [ITS]), but their sensitivity to detect positive subclinical samples has been questioned, usually attributed to the low bacterial numbers in the blood, resulting in low DNA extraction yield (Engvall et al. 2003, Rampersad et al. 2005, Ayllon et al. 2012). Therefore, more sensitive assays, such as nested or real-time PCR, have been required for the detection of Bartonella in samples with low DNA concentrations (Ciervo and Ciceroni 2004, Rampersad et al. 2005, Mietze et al. 2011). Furthermore, high-resolution melt (HRM) real-time PCR analysis has provided a sensitive, closed-system and rapid assay for graphic discrimination between several Bartonella species (Morick et al. 2009). To date, only a few studies have applied real-time PCR assays for the detection of Bartonella prevalence, but besides one that included rodent-associated Bartonella spp. only (Morick et al. 2009), none have applied HRM real-time PCR for discrimination among all feline-associated Bartonella spp.
The present study investigated the prevalence of Bartonella spp. in blood of domestic and stray cats from Israel, and compared culture-isolation with HRM real-time PCR as screening methods for the diagnosis of feline bartonellosis.
Materials and Methods
Cats sampling and data collection
From January, 2008, to November, 2010, EDTA–blood samples were collected under sterile conditions from stray and domestic cats in private clinics from Tel Aviv and Mishmar Hasharon, Israel. After collection, samples were chilled and transported to the laboratory of the Koret School of Veterinary Medicine and kept under freezing (−20°C) until further analyzed. The cat gender, categorical age (kittens, <1 year; young adult, 1–4 years; mature, >4 years), flea infestation status, and the city of origin were recorded. The study was approved by the Hebrew University Medical Faculty Institutional Animal Care and Use Committee (MD12134612).
Bacterial isolation and molecular screening of cultured colonies
EDTA–blood samples were defrosted and homogenized at room temperature. Subsequently, 200 μL were plated on chocolate agar and incubated at 37°C in a 5% CO2 atmosphere for up to 8 weeks. All agar plates were checked weekly during the incubation period for the presence of typical Bartonella cultures. The latter were diagnosed as small, white-creamy in color, rough, and dry colonies. Morphological evaluation was done to each positive culture, and distinct colonies were selected. Colonies with different apparent morphology were recultured to allow detection of co-infections with several different Bartonella species.
DNA was extracted from the colonies using a DNA-extraction kit (Illustra Tissue Mini Spin kit, GE Healthcare, Little Chalfont, UK) according to the manufacturer's instructions. Colonies were confirmed to be Bartonella positive by conventional PCR amplification targeting a 379-bp fragment of the Bartonella gltA sequence using primers Bhcs.781p (GGGGACCAGCTCATGGTGG) and Bhcs.1137n (AATGCAAAAAGAACAGTAAACA) (Norman et al. 1995). The gltA-PCR reactions were carried out in a 25-μL final volume using PCR-Ready High Specificity ready mix (Syntezza Bioscience Ltd, Jerusalem, ISR) containing 1 μL of 10 μM solution of each primer, 21 μL double-distilled water (DDW), and 2 μL of each extracted DNA sample. The amplification products were obtained by the following protocol: 2 min at 94°C followed by 35 cycles of 30 sec at 94°C, 30 sec at 54°C, and 1 min at 72°C, and a final step of 5 min at 72°C. The PCR products were run on 1.5% agarose gels and visualized under ultraviolet (UV) light to determine any positive amplification. All positive products were confirmed by sequencing as described below. Additionally, all DNA samples extracted from colonies were further screened for Bartonella ITS, rpoB, and 16S rRNA loci by HRM real-time PCR, in parallel with DNA samples extracted from whole- blood, as described below.
HRM real-time PCR whole-blood DNA screening
Genomic DNA was extracted from 50 μL of EDTA blood of each cat, using a DNA-extraction kit (BiOstic Bacteremia DNA Isolation Kit; MO BIO Laboratories, Inc., Carlsbag, CA). DNA was obtained in 50 μL of elution buffer. For quality assurance, a negative Bartonella sp. blood sample was used as extraction control in the preparation.
The molecular screening for Bartonella spp. of the whole-blood DNA was assessed by real-time PCR followed by HRM analysis, targeting the 16S–23S ITS. A 190-bp fragment was amplified using primers 321s (AGATGATGATCCCAAGCCTTCTGG) and H493as (TGAACCTCCGACCTCACGCTTATC) (Maggi and Breitschwerdt 2005). The ITS real-time PCR reactions were carried out in a 20 μL final volume containing 1 μL of 10 μM solution of each primer, 0.6 μL of 50 μM solution of Syto9 (Invitrogen, CA), 3.4 μL of DDW, 10 μL of MAXIMA Hot-Start PCR Master Mix 2X (Thermo Scientific, Surrey, UK), and 4 μL of each genomic DNA. The amplification products were obtained by the following protocol: 4 min at 95°C followed by 50 cycles of 5 s at 95°C, 15 s at 60°C (fluorescence acquisition on HRM channel), and 1 s at 72°C. The melt, hybridization, and HRM phases were performed as previously described (Morick et al. 2009). Any amplification with a threshold cycle (Ct) value lower than 40 cycles was considered as a positive result. The system was standardized using positive DNA controls. Additionally, a Bartonella- positive DNA, a Bartonella-negative DNA, and a nontemplate DNA control were used in each run. All reactions were carried out using the Rotor Gene 6000 cycler (Corbett Research, Sydney, Australia). All positive amplicons were sequenced as described below.
All whole-blood DNA samples that were positive by the ITS real-time PCR were later screened for the rpoB and the 16S rRNA gene fragments by HRM real-time PCR. A 210-bp rpoB gene fragment was amplified using primers 800r (GATCTAAATCTTCTGTTGCACG) and 600f (GAAAATGATGATGCGAATCG) (Morick et al. 2009). The rpoB real-time PCR was performed in a 20 μL reaction volume containing 0.5 μL of 10 μM solution of each primer, 0.6 μL of 50 μM solution of Syto9, 4.4 μL DDW, 10 μL of MAXIMA Hot-Start PCR Master Mix 2X, and 4 μL of each genomic DNA. The amplification conditions were set as previously described (Morick, et al. 2009). The third HRM real-time PCR reaction amplified a 296-bp 16S rRNA gene fragment, using primers p24E (GGAATTCCCTCCTTCAGTTAGGCTGG) and p12B (CGGGATCCCGAGATGGCTTTTGGAGATTA) (Relman et al. 1990). The reagent concentrations used for the 16S rRNA gene fragment amplification were the same as for the ITS real-time PCR (described above). The amplification products were obtained by the following protocol: 4 min at 95°C followed by 50 cycles of 5 s at 95°C, 30 s at 60°C (fluorescence acquisition on HRM channel), and 15 s at 72°C. Melt and HRM analyses for the rpoB and 16S assays were performed as those used for the ITS real-time PCR (described above).
All primer sequences were verified in silico using MEGA alignment software (version 5.05, The Biodesign Institute, Tempe, AZ) for the amplification of the feline Bartonella spp. DNA sequences (GenBank database for B. henselae, B. clarridgeiae, B. koehlerae, B. quintana, and B. bovis).
Sequencing
All positive PCR products were purified using a PCR purification kit (Exo-SAP, NEB; New England Biolabs, Inc., Ipswich, MA) and subsequently sequenced using BigDye Terminator cycle sequencing chemistry from Applied Biosystems ABI 3700 DNA Analyzer, and the ABI Data collection and Sequence Analysis software (ABI, Carlsbag, CA). Further analysis was done using the BioEdit Sequence Alignment Editor software, version 7.1.3.1 (Ibis Biosciences, Carlsbag, CA).
Statistical Analysis
Statistical analysis of the associations between Bartonella infection rates in stray and domestic cats and the distributions of Bartonella species within cat populations were performed using the Pearson chi-squared test or Fisher exact test. Significance was defined as p<0.05.
Results
Cats
A total of 334 EDTA cat blood samples (179 stray cats and 155 domestic cats) were collected from 18 cities or villages and their surroundings in central and northcentral Israel. Of all 179 stray cats, 99 (57%) were females and 76 (43%) were males (gender was not recorded for 4 cats). One hundred and sixty-nine (94.4%) cats were infested with fleas at the moment of the collection. The range of categorical ages was distributed as 37 (21%) kittens, 116 (66%) young adults, and 23 (13%) mature cats (age unknown for 3 cats). Of the 155 domestic cats, 29 (36%) were females and 52 (64%) were males (gender was not recorded for 74 cats). Categorical ages were distributed as 3 (6%) kittens, 34 (65%) young adults, and 15 (29%) mature cats (unknown age for 103 cats). No information on the flea infestation status was recorded in the domestic cats group.
Bartonella spp. identification
Bartonella spp. were detected in 25.1% (84/334) of all cats, by one or both detection methods (culture isolation and whole-blood DNA HRM ITS real-time PCR assays). The prevalence of infection with Bartonella spp. was 30.7% (55/179) in the stray cats and 18.7% (29/155) in the domestic cats, being significantly higher in the stray cat population (p=0.012, Pearson chi-squared test) (Table 1).
ND, not detected.
Significant difference.
Overall, the Bartonella spp. identified in both cat populations were B. henselae, B. clarridgeiae, and B. koehlerae (15.6% [52/334], 8.1% [27/334], and 3.6% [12/334], respectively). The distribution of these species varied significantly between the cat populations (p=0.019, Pearson chi-squared test) (Fig. 1). The prevalence of B. clarridgeiae and B. koehlerae was significantly higher in the stray cats (12.3% [22/179] and 5.6% [10/179], respectively), compared to the domestic cats (3.2% [5/155] and 1.3% [2/155)] respectively) (p=0.002 and 0.035, respectively; Pearson chi-squared test). No significant difference was noticed in the prevalence of B. henselae between the stray and the domestic cats (15.6% [28/179] and 15.5% [24/155], respectively) (p=0.968, Pearson chi-squared test) (Table 1). Neither B. bovis nor B. quintana was detected in any cat sample.
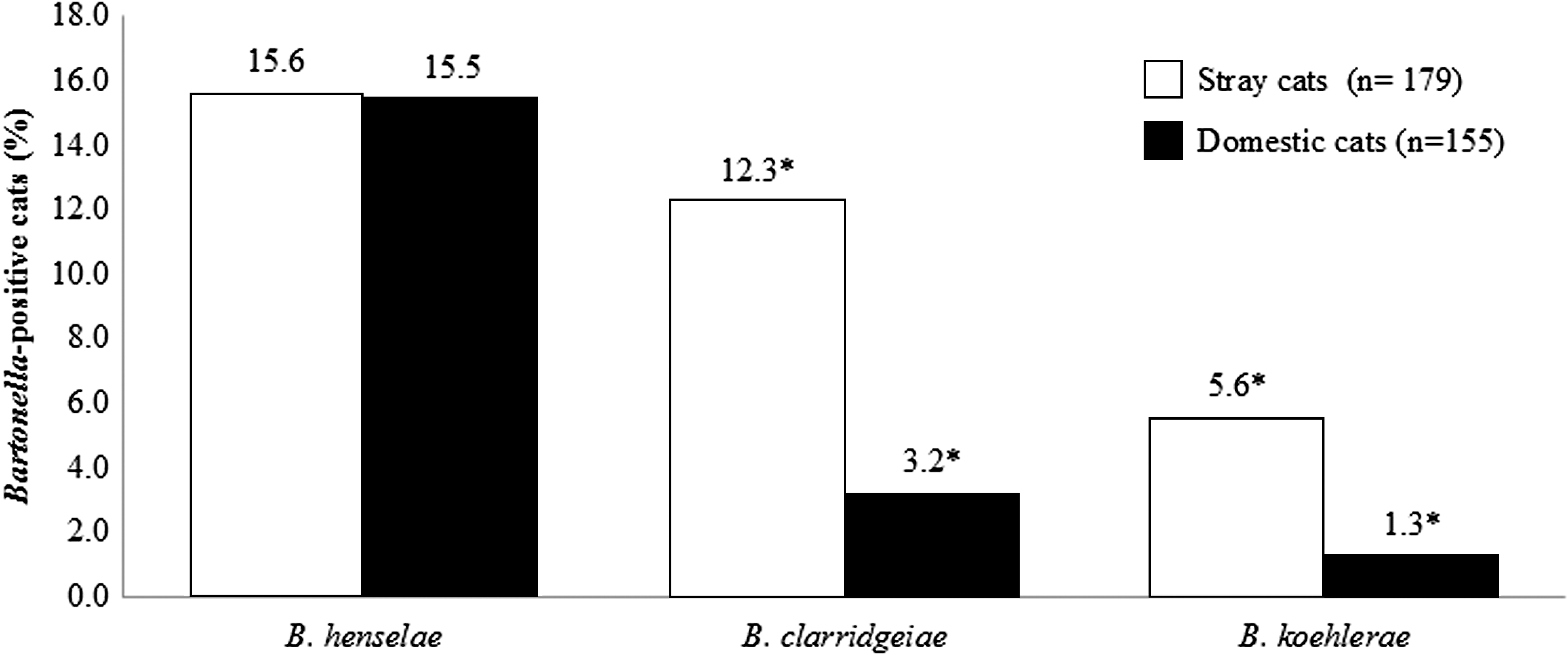
Distribution of Bartonella species within Bartonella-positive cats according to the cat population (stray versus domestic). (*) Significantly different (p<0.05).
Coinfection was diagnosed in seven (2.1%) cats: B. henselae and B. clarridgeiae (3 cats), B. henselae and B. koehlerae (3 cats), and B. clarridgeiae with B. koehlerae (1 cat). In addition, a domestic cat coinfected with B. henselae and B. koehlerae was infected with a third Bartonella sp., designated as Bartonella sp. FG 4-1 (100% sequence similarity; GenBank accession number AB529960.1). Five of the coinfected cats were stray, and only two were domestic, but there was no significant difference between percentages of coinfection between the cat populations, 2.8% (5/179) and 1.3% (2/155), respectively (p=0.339, Pearson chi-squared test).
Cat population analysis
Stray cats
No significant difference in the infection rate was noticed between the female and male stray cats (30% and 32%, respectively; p=0.870, Fisher exact test). All Bartonella-positive cats but one were found to be infested with fleas at the time of sampling. In all, 38% (14/37) of the kittens and 32% (37/116) of the young adult stray cats were infected with Bartonella spp., showing no significant difference between these age groups (p=0.550, Fisher exact test), whereas stray kittens had significant higher prevalence than mature stray cats (13%; 3/23) (p=0.045, Fisher exact test). There was no significant difference in Bartonella infection rates between young adult cats and mature stray cats (p=0.080, Fisher exact test).
Domestic cats
No significant difference in the infection rate was noticed between the female and male domestic cats (17% versus 21%, respectively; p=0.776, Fisher exact test). No significant difference in infection rate between the different age groups was detected (p>0.05; Fisher exact test).
Screening method performance
The whole-blood DNA screening by the HRM ITS real-time PCR assay detected 94.0% (79/84) of all Bartonella-positive feline samples, whereas 35.7% (30/84) were detected by bacterial culture isolation. Of the former, B. henselae was detected in 77% (40/52), B. clarridgeiae in 100% (27/27), and B. koehlerae in 83% (10/12), and two samples were confirmed only as Bartonella sp. by sequencing (>90% sequence similarity with Bartonella genus, later confirmed to be B. henselae by sequencing of the rpoB gene segment). On the other hand, all DNA samples extracted from the positive-cultured samples were confirmed to be B. henselae by sequencing, and one plate was confirmed to be coinfected with colonies of B. koehlerae and Bartonella sp. FG 4-1. B. clarridgeiae was not detected in any DNA sample extracted from the cultured colonies. Five of the positive-cultured samples were negative through whole-blood DNA HRM ITS real-time PCR. Moreover, two samples that were positive for B. henselae by culture isolation and confirmed by gltA PCR on DNA extracted from the colonies were found to be co-infected with B. koehlerae through whole-blood DNA HRM ITS real-time PCR and sequencing.
The ITS HRM real-time PCR analysis showed a high discriminatory power between B. henselae, B. clarridgeiae, and B. koehlerae (Fig. 2). The range of the HRM melting peaks varied as follows: 82.2–83.0°C, 83.2–83.9°C, and 84.0–84.4°C for B. clarridgeiae, B. henselae, and B. koehlerae, respectively. Each graphical HRM pattern matched 100% with sequencing results obtained for each of the three respective Bartonella spp.
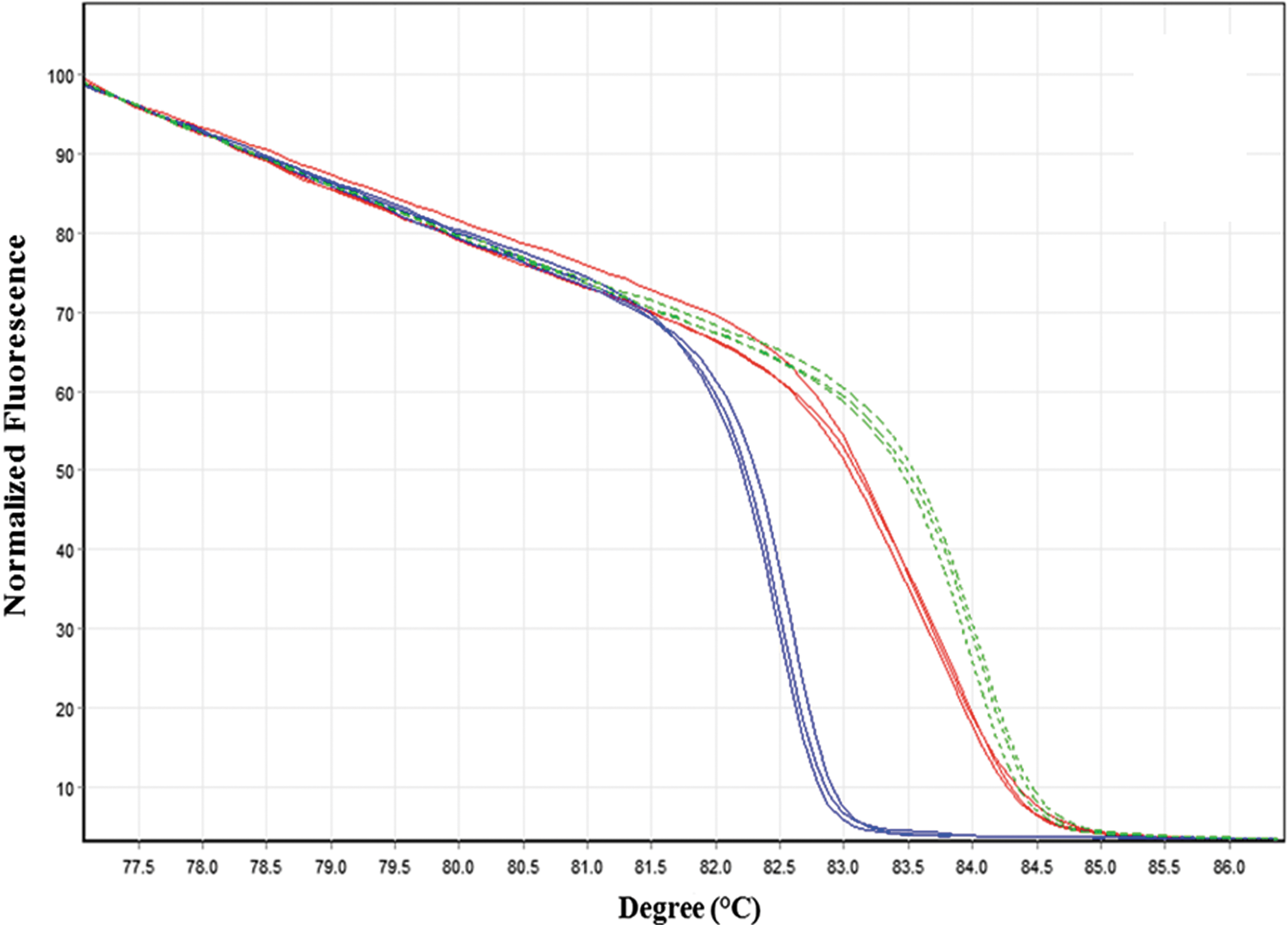
Normalized high-resolution melt (HRM) real-time PCR analysis for identification of internal transcribed spacer (ITS) fragments of B. henselae (solid red line), B. clarridgeiae (solid blue line), and B. koehlerae (dashed green line). Each Bartonella species is represented by three sequence-confirmed positive samples. Color images available online at
Using the whole-blood DNA rpoB and 16S HRM real-time PCR assays, Bartonella DNA was detected in 65.5% (55/84) and 71.4% (60/84) of all whole-blood DNA positive cat samples, respectively. Both real-time PCR assays amplified B. henselae–, B. clarridgeiae–, and B. koehlerae–specific DNA sequences. Additionally, two of the positive Bartonella cultures that were not amplified by whole-blood DNA HRM ITS real-time PCR were confirmed positive when targeting one of these two genes. Furthermore, using these complementary HRM real-time PCR assays, co-infection was detected in four additional cases. Notwithstanding, HRM analysis of rpoB and 16S gene fragments showed a low discriminatory power for the three feline Bartonella species detected. The range of the HRM melting peaks for rpoB fragments varied as follows: 83.8–84.6°C, 84.0–84.1°C, and 83.7–84.5°C, and for 16S fragments from 88.2–88.6°C, 88.0–88.4°C, and 88.2–88.6°C for B. henselae, B. clarridgeiae, and B. koehlerae, respectively.
Overall, confirmation by DNA sequencing for B. henselae, B. clarridgeiae, and B. koehlerae showed sequence similarity percentages ranging from 98–100% for ITS (GenBank accession numbers JQ638927.1, DQ683194.1, and AF312490.1, respectively), 97–100% for rpoB (GenBank accession numbers JN646673.1, FN645454.1, and FJ832089.1, respectively), and 99–100% for 16S fragments (GenBank accession numbers JN646650.1, EU571939.1, and AF076237.1, respectively). Furthermore, positive Bartonella colonies DNA showed a gltA sequence similarity percentage that ranged from 99–100% for B. henselae (GenBank accession number FJ492803.1).
Discussion
This study demonstrates a high overall molecular prevalence (25.1%) of Bartonella infection in cats from 18 cities and villages in central and northcentral Israel. Recent studies that included both stray and domestic cats have reported lower infection rates in the overall feline populations in Spain, China, and Thailand ranging from 0.3% to 15.1% (Inoue et al. 2009b, Yuan et al. 2011, Ayllon et al. 2012). However, higher rates were reported from South Korea, reaching 40.4% of the total cat population (Kim et al. 2009). In our study, the infection rate was significantly higher in stray (30.7%) than domestic cats (18.7%) (p=0.012). Such findings were also described in Spain, China, and Thailand, and varied from 0.7% to 23.9% in stray cats as compared to 0% to 11.1% in domestic cats (Inoue et al. 2009b, Yuan, et al. 2011, Ayllon et al. 2012). The reported prevalence in the South Korean stray cats was 43.9% compared to 33.3% in domestic cats (Kim et al. 2009). The latter prevalence was higher compared to recent reports from Brazil (4.3%), Germany (16.6%), and Thailand (17%), where domestic cats only were included in the studies (Mietze et al. 2011, Assarasakorn et al. 2012, Bortoli et al. 2012). Additionally, infection rates in stray kittens in our study were significantly higher than mature cats, as has been previously reported in domestic cats from the United States and Germany (Chomel et al. 1995, Mietze et al. 2011). On the contrary, no significant differences between any categorical ages were detected in the domestic cats in this study, probably due to the relatively small number of cats with available data in this group.
Although, B. henselae, B. clarridgeiae, and B. koehlerae were isolated in the past from stray cats in Israel (Avidor et al. 2004), the prevalence of these species was not determined in either stray or domestic cats from this country to date. Our study indicates an uneven distribution of the three detected Bartonella species between the feline populations (Fig. 1), where B. clarridgeiae and B. koehlerae were significantly more prevalent in stray than domestic cats (p=0.016). The overall infection rate for B. clarridgeiae (8.1% in the general cat population) was higher than recent reports from South Korea, Germany, Thailand, China, and Spain, that varied from 0.15% to 4.5% (Kim et al. 2009, Mietze et al. 2011, Yuan et al. 2011, Assarasakorn et al. 2012, Ayllon et al. 2012). Moreover, our study detected a higher than expected prevalence rate of B. koehlerae within the Israeli feline population (3.6% in the general cat population and 5.6% in stray cats). In fact, B. koehlerae had been reported in only a few cases and reports worldwide, since it was first described in two domestic cats from the United States (Droz et al. 1999) and more recently in 1.3% of domestic cats screened from Thailand (Assarasakorn et al. 2012). The same percentage was detected in domestic cats in the present study. Additionally, our study did not detect any cat infected with B. quintana or B. bovis, suggesting that these species are not present or that their prevalence is very low in the Israeli feline populations. Interestingly, an equal distribution of B. henselae was detected in stray and domestic cats in the current study (15.6% and 15.5%, respectively), reflecting the extensive spread of B. henselae, and probably their flea vector, in cats from Israel.
Coinfections of cats, with B. henselae and B. clarridgeiae have been reported from other countries in varied percentages (0.7–2.9%) (Maruyama et al. 2001, Rampersad et al. 2005, Yuan et al. 2011, Assarasakorn et al. 2012). Additionally, a domestic cat coinfected with B. henselae and B. koehlerae was recently reported from Thailand (Assarasakorn et al. 2012). The present study detected seven cases (2.1%) of coinfection with the three above-mentioned feline Bartonella spp. in all possible combinations, including a cat with B. clarridgeiae and B. koehlerae. Furthermore, the detection of a rodent-associated Bartonella (Bartonella sp. FG 4-1) in one domestic cat coinfected with B. henselae and B. koehlerae deserves a special attention. Although, this coinfecting genotype (Bartonella sp. FG 4-1) was isolated by culture, it could only be detected through the amplification of the rpoB gene fragment by real-time PCR (100% sequence similarity, accession number AB444987.1 GenBank) and not through targeting the other loci. Bartonella sp. FG 4-1 was initially detected from a bushy-tailed jird (Sekeetamys calurus) from Egypt (Inoue et al. 2009a), a common rodent species found also in Israel, and from a gerbil (Gerbillus nanus) from the Negev desert, Israel (Morick et al. 2011). Although this genotype host was a domestic cat, it was an indoor–outdoor cat, suggesting the possibility of hunting and acquiring this species from an infected rodent via a route yet to be identified.
Bacterial culture of suspected blood had been the preferred identification method for Bartonella infections; however, it is laborious, time consuming, and often insensitive. For this reason, new molecular detection assays have been developed to overcome these limitations. A recent study demonstrated 2.2% positive cultures in cats from Germany; however, when DNA extracted from blood was subjected to real-time PCR, 16.6% of the same cats were diagnosed to be Bartonella positive (Mietze et al. 2011). Additionally, a recent study presented a novel real-time PCR assay targeting the ssrA RNA gene (tmRNA), which also proved to be more sensitive than bacterial culture in the detection of elk and cattle Bartonella spp. (Diaz et al. 2012). However, due to the fact that it is a new target gene in Bartonella diagnosis, it has a limited sequence database in GenBank, which limits its application for prevalence and comparative studies at the current stage. Alternative enrichment-liquid culture media have also been developed to improve the detection of Bartonella-positive samples (Maggi et al. 2005, Duncan et al. 2007, Riess et al. 2008, Bai et al. 2010). Nevertheless, the present study compared culture-isolation by the traditional chocolate agar media with HRM real-time PCR assays using common targeted Bartonella loci (ITS, rpoB, and 16S rRNA). Similarly, the use of the three HRM real-time PCR assays together was more sensitive than bacterial isolation (81/84 [96.4%] versus 30/84 [35.7%], respectively). The ITS HRM assay alone was found to be highly sensitive and specific, diagnosing 94.0% (79/84) of all Bartonella-positive feline samples, and presented the additional advantage of direct and immediate identification of the three feline Bartonella species through the characteristic curve of the ITS HRM assay (Fig. 2). Recently, HRM real-time PCR using the rpoB fragment was successfully described for the detection and discrimination of several different rodent-associated Bartonella spp. (Morick et al. 2009). However, the rpoB HRM rodent assay showed overlap in melting curves for the three feline Bartonella spp. amplicons identified in this study, which prevented their discrimination by visualizing the HRM graphical patterns. Similar results were obtained by the 16S HRM real-time PCR assay. In silico analysis showed that the rpoB 600f primer presented two mismatches in the 3′ end for the B. clarridgeiae rpoB sequence that probably influenced in the detection of only six out of 27 positive samples containing DNA of this species. This was not surprising because primer mismatches in the 3′ end are known to cause an increase in the Ct value in real-time PCR amplification (Whiley et al. 2008). Interestingly, this primer mismatch allowed the detection of other Bartonella spp. that were present in the same sample, probably as a result of a primer-annealing competitive event that favored the detection of one of the sequence variants (Whiley et al. 2008). Moreover, a previous study showed that in an experimental coinfected sample with two Bartonella spp., a double-peak pattern can be detected in the melting curve through a single real-time assay (Ciervo and Ciceroni 2004). However, in the present study, only three of all coinfected samples showed this double-peak phenomenon. On the other hand, three Bartonella-positive samples were detected only through culture, indicating the possibility of the presence of PCR inhibitors or low DNA extraction yield, preventing the molecular detection of Bartonella DNA, in those particular cases. Our results indicate that the combination of real-time PCR and isolation methods is recommended in the diagnosis of feline Bartonella spp. as they complement each other and decrease the number of false-negative results in a suspected population.
The citrate synthase gene (gltA) has been used extensively for identification of Bartonella genotypes, due to its potent discriminatory power (La Scola et al. 2003). In the present study, it was found useful when DNA extracted from bacterial colonies was used. Conversely, it could not be applied for the HRM real-time PCR when whole-blood DNA samples were screened, due to a nonspecific amplification (of an ∼800-bp amplicon). This limitation has been previously reported by others (Avidor et al. 2004, Colborn et al. 2010, Diaz et al. 2012). However, in the present study, it was not possible to determine the origin of the cross-reactivity, probably attributed to a multiple same-size fragment amplification that affected the reading by the sequencer.
In conclusion, this study demonstrated that B. henselae, B. clarridgeiae, and B. koehlerae are prevalent in Israeli cats. Infection rates with B. clarridgeiae and B. koehlerae were shown to be significantly higher in stray than domestic cats, whereas B. henselae was highly and equally distributed among both domestic and stray cat populations. Altogether, our results reflect the widespread of the three major feline zoonotic Bartonella species within stray and domestic cat populations in Israel, highlighting their potential zoonotic and public health importance. Finally, this study demonstrated that HRM real-time PCR assay targeting the Bartonella ITS region is a sensitive, specific, and rapid assay for the diagnosis of feline Bartonella spp. and suggests the importance of applying more than one diagnostic method to overcome false-negative results when diagnosing Bartonella spp.
Footnotes
Acknowledgments
This study was funded by The Ministry of Foreign Affairs, The Hague, The Netherlands, through a grant to the Dutch Friends of the Hebrew University (NVHU). The authors gratefully acknowledge Dr. Gal Melamed from The Veterinary Clinic at Yakum, Israel, and Dr. Eyal Nachmias from The Veterinary Clinic at Mishmar Hasharom, Israel for their assistance in sample collection.
Author Disclosure Statement
No competing financial interests exist.