
Abstract
Rickettsia felis is an obligate, intracellular, Gram-negative bacterium and a member of the transitional group rickettsiae. This bacterium has been shown to grow in vitro in amphibian, tick, and mosquito cell lines. Here, we present data to show the growth of R. felis strain LSU in Drosophila melanogaster S2 cells, an embryonic, hemocytic cell line with phagocytic properties. R. felis LSU was isolated from Ixodes scapularis E6 (ISE6) cells and used to infect S2 cells, grown at 25°C. By 19 days postinfection, the S2 cells were 100% infected with R. felis as determined by Acridine Orange and Diff-Quik staining. A species-specific R. felis qPCR assay was used to demonstrate that the kinetics associated with the S2 cell culture infection involved a lag/adaptation phase, followed by continued growth to 20 days postinfection. Moreover, R. felis organisms were observed in the S2 cells using transmission electron microscopy and a polyclonal antibody against spotted fever rickettsiae. The ability to use D. melanogaster S2 cells for growing rickettsial agents is a useful tool owing to the ease of manipulation of the S2 cultures and the wide-ranging possibility of Drosophila resources available for future studies.
Introduction
Rickettsia felis
Human infection with R. felis causes flea-borne spotted fever (also known as cat-flea typhus and R. felis rickettsiosis); it was first reported in the United States in 1994 (Schriefer et al. 1994). Today, R. felis is considered to be an emerging infectious disease, and human cases have been reported in North America, South America, Europe, Africa, Asia, and New Zealand/Australia (Renvoise et al. 2009, Williams et al. 2011, Lim et al. 2012). Symptoms of the disease are similar to other rickettsioses and can include fever, rash, headache, muscle pain, fatigue, eschar at the site of the flea bite, and other flu-like symptoms. No deaths from human infection with R. felis have been formally reported (Hun and Troyo 2012), and doxycycline as a treatment option has been used with success (Hun and Troyo 2012). Due to the worldwide emerging nature of this pathogen, the cosmopolitan distribution of its recognized vector and the possibility of potential transmission by a milieu of other arthropod vectors, systems for optimally establishing, propagating, and maintaining isolates of R. felis are needed.
Growth of R. felis in vitro has been accomplished using several different cell lines. Published reports demonstrate that the bacteria can be successfully propagated in mosquito cells (cell lines Aa23, Sua5B, and C6/36) (Horta et al. 2006, Sakamoto and Azad 2007), tick cells (cell line ISE6) (Pornwiroon et al. 2006), and amphibian cells (cell line XTC-2) (Raoult et al. 2001, La Scola et al. 2002). It has been demonstrated that mammalian cell lines (Vero and L929), particularly with special medium supplementation, can also be used to grow the organism (Saisongkorh et al. 2011). A common characteristic to the successful propagation of R. felis in the aforementioned cell lines is incubation at lower temperatures (e.g.,<31°C). We hypothesized that R. felis strain LSU could be successfully grown in Drosophila melanogaster S2 cells. S2 cells are an embryonic-derived hemocytic cell line with phagocytic properties and functional immune signaling (Baum and Cherbas 2008). Drosophila cell lines have been used to study other intracellular pathogens such as Ehrlichia chaffeensis (closely related to rickettsiae) (Luce-Fedrow et al. 2008), Rickettsia parkeri (Serio et al. 2010, Reed et al. 2011), Listeria monocytogenes (Cheng and Portnoy 2003), Francisella tularensis (Santic et al. 2009), and Chlamydia trachomatis (Elwell and Engel 2005).
The ease of maintenance and the ability to culture S2 cells at low temperatures makes them an attractive tool to use for studying rickettsial pathogens. Moreover, approximately 50% of D. melanogaster genes have mammalian homologs (Bernards and Hariharan 2001), and loss-of-function mutants are currently available for approximately 53% of D. melanogaster genes (Igboin et al. 2012). These features of the Drosophila system could prove to be valuable tools for analyzing rickettsial host–pathogen relationships in the future. Here, we present data demonstrating the successful propagation and maintenance of R. felis LSU in S2 cells using molecular and cytological techniques. The growth of R. felis in S2 cells helps to demonstrate the utility of S2 cells for studying rickettsial pathogens and will ultimately contribute to enhancing the body of knowledge concerning the growth and biological characteristics of R. felis in its arthropod hosts.
Materials and Methods
Maintenance of cell lines
The Vero cell line (American Type Culture Collection [ATCC], Manassas, VA) was maintained at 37°C, 5% CO2 in Dulbecco modified Eagle medium (DMEM) (ATCC) containing 5% fetal bovine serum (FBS; Atlanta Biologicals, Lawrenceville, GA). The S2 cell line (Drosophila Genomics Resource Center, Bloomington, IN) and C6/36 cell line (gift of Dan Ewing, Naval Medical Research Center, Silver Spring, MD) were cultivated at 25°C in Schneider's Drosophila medium (Gibco/Invitrogen, Carlsbad, CA) supplemented with 5% FBS. No antibiotics were used for cell culture.
Initial infections and time course infections in S2, C6/36, and Vero cells
R. felis LSU was isolated from the ISE6 cells by low- and high-speed centrifugation. The ISE6 cells were removed from the flask and mechanically disrupted to release the bacteria by shaking with glass beads. The culture was then centrifuged at 600× g for 20 min. The resulting supernatant was subsequently transferred to a sterile conical tube and centrifuged at 15,000 rpm for 20 min. The final bacterial pellet was resuspended in 2 mL of sterile Schneider Drosophila medium (containing 5% FBS) and used to inoculate two 25-cm2 flasks (Corning, Tewksbury, MA) of D. melanogaster S2 cells. The S2 cells were monitored for the growth of R. felis by cytological stain and R. felis qPCR assay as described below. When cells reached 90–100% infection, as visualized by Acridine Orange and Diff-Quick staining, aliquots were used to establish new infections in uninfected S2 cells and/or frozen at −80°C using Recovery Cell Culture Freezing Medium, per the manufacturer's instructions (Gibco/Invitrogen).
For time-course infections in S2, C6/36, and Vero cells, cells were plated at a density of 1×106 cells/mL in 25-cm2 flasks (Corning) 24 h prior to infection with R. felis. For time-course infections in S2 cells only, between 1 and 3 mL of a 90–100% infected S2 cell culture (as determined by Acridine Orange and Diff-Quik staining) was transferred to the newly seeded S2 cells cultures. For simultaneous time course infections in the S2, C6/36, and Vero cells, R. felis was isolated from a 90–100% infected flask of S2 cells by low- and high-speed centrifugation, as previously described. The final bacterial pellet was resuspended in 4 mL of sterile phosphate-buffered saline (PBS); 1 mL was used to infect each flask of S2, C6/36, or Vero cells. DNA was extracted from a 1-mL aliquot of the infected S2 cells used to initiate infection in the uninfected S2 cells or from the cell-free preparation used to initiate simultaneous infections in the S2, C6/36, and Vero cells. This DNA preparation served as a representation for the number of DNA copies present at a time point of 0 days postinfection (dpi) in the time-course infection experiments. At selected time points in all time-course infection experiments, cells/medium were removed by drawing a pipette across the bottom of the flask(s); 1 mL of the cells/medium was used for DNA extraction and another 1 mL of the cells/medium was used for cytological staining as described below. Fresh medium was used to replace that which was removed with the cells for DNA extraction and staining each time a sample was taken from the flask(s).
Infections in all three cell lines were also visualized at various time points using Diff-Quik stain (Andwin Scientific, Schaumburg, IL) and Acridine Orange stain (Amersham Biosciences/GE Healthcare, Piscataway, NJ). Cells were prepared on glass slides following centrifugation of 1 mL of cells sampled from the flask and resuspension in 20–50 μL of sterile PBS; 5 μL of cells were then heat fixed to the slides. Slides were stained with the Diff-Quik kit according to manufacturer's instructions. For Acridine Orange staining, slides were dried at room temperature, flooded with Acridine Orange for 2 min, and then rinsed in distilled water. Images were viewed with an Olympus BX43 microscope and DP72 camera (Olympus, Center Valley, PA). R. felis infections were quantitated by assessing the number of cells containing at least one rickettsial organism by randomly scoring fields of cells for the presence or absence of one or more intracellular rickettsial organisms. A total of 100–150 cells were randomly scored for each time point of each experiment.
Transmission electron microscopy
Infected (17–20 dpi) S2 cells and uninfected S2 cells were prepared for transmission electron microscopy (TEM). Cells were removed from flasks by scraping with a cell scraper (Fisherbrand, Pittsburgh, PA), transferred to 50-mL conical tubes, and centrifuged for 5 min at 1200 rpm. Supernatants were discarded, and the samples were immersion fixed in fixative containing 4% formaldehyde/1% glutaraldehyde in 0.1 M sodium phosphate buffer. The samples were fixed for 1 h, washed three times with 0.1 M sodium phosphate buffer (15 min per wash), and then postfixed with 1% Osmium Tetroxide I 0.1 M sodium phosphate buffer for 1 h. The images were collected on a JEM 100 CX II transmission electron microscope (JEOL, Japan).
Immunocytochemistry
Immunocytochemistry was performed on R. felis–infected and uninfected S2, C6/36, and Vero cells. The cells were prepared on glass slides following centrifugation of 1 mL of cells and resuspension in 20–50 μL of sterile PBS; 5 μL of cells were air dried on the slide(s), fixed with acetone, dried for 10 min, and outlined using a Pap Pen (The Binding Site Inc., San Diego, CA). Samples were placed in PBS containing 0.01% Tween-20 for 5 min and then blocked with PBS containing 10% normal goat serum for 30 min at 37°C. Samples were washed in PBS for 5 min and incubated with primary antibody (polyclonal rabbit–anti-spotted fever–R. rickettsii Sheila Smith, produced by Richards Lab; 1:100 dilution) for 24 h in the dark at 4°C or with normal rabbit serum as a control. The slides were washed with PBS for 5 min and were incubated for 1 h in the dark at room temperature with a 1:500 dilution of Alexa Fluor 488 goat anti-rabbit immunoglobulin G (IgG) (H+L) (no. A11008, Invitrogen/Molecular Probes). Samples were washed in PBS for 5 min and then in distilled water for 5 min before viewing. Images were viewed with an Olympus BX43 microscope and DP72 camera (Olympus).
Determination of infection by qPCR
R. felis infections in S2, C6/36, and Vero cells were assessed by a R. felis qPCR assay as previously described (Henry et al. 2007). All qPCR reactions were performed using the Platinum qPCR UDG Supermix kit (Invitrogen) in a Cepheid Smart Cycler. The number of DNA copies of R. felis was normalized to the housekeeping genes β-actin (for Vero cells) (Luce-Fedrow et al. 2011), Drosophila ribosomal protein 15a (for S2 cells) (Schneider and Shahabuddin 2000), and Aedes albopictus ribosomal protein S7 (for C6/36 cells). A. albopictus rpS7 was detected using forward primer, 5′-ACGTCGTGTTCATTGGTGAG; reverse primer, 5′-CGA CCTTGTGTTCAATGGTG; and probe FAM, 5′-ATGCC ATCCTTGAGGATCTG-3′BHQ-1. Cycling conditions for rpS7 were as follows: 50°C for 120 s, 95°C for 120 s, followed by 45 cycles of 95°C for 15 s, and 58°C for 30 s. Negative controls consisting of uninfected cells and reactions devoid of template were also subjected to our R. felis qPCR assay with each qPCR run. No rickettsial copies were detected in our negative controls.
Results
S2 cells support the growth of R. felis
To determine if R. felis could be propagated in D. melanogaster S2 cells, we used R. felis strain LSU isolated from ISE6 cells to inoculate uninfected S2 cells. Infections were detected and monitored by qPCR, cytological staining with Acridine Orange and Diff-Quik stains, TEM, and immunocytochemistry. Initial growth of the bacteria in the S2 cells was confirmed using Acridine Orange and Diff-Quik stains. It was initially found that by 17–20 dpi, 90–100% of the S2 cells were infected with R. felis LSU. To better understand the kinetics of infection, three independent time-course infections were set up in the S2 cells and monitored at various time points for the presence of rickettsial organisms within the S2 cells (Fig. 1) and for the number of DNA copies present at each time point (Fig. 2). When S2 cells were scored for the presence or absence of at least one intracellular rickettsial organism at selected time points, we observed an increase in the number of cells that contained at least one organism from 1–20 dpi (Fig. 1). At 1 dpi, approximately 20% of the S2 cells contained at least one organism, by 8 dpi upward of 50% of the cells contained at least one rickettsial organism, and by 20 dpi 100% of the S2 cells were infected with R. felis (Fig. 1). Similar infection kinetics were observed across the three independent infection experiments.
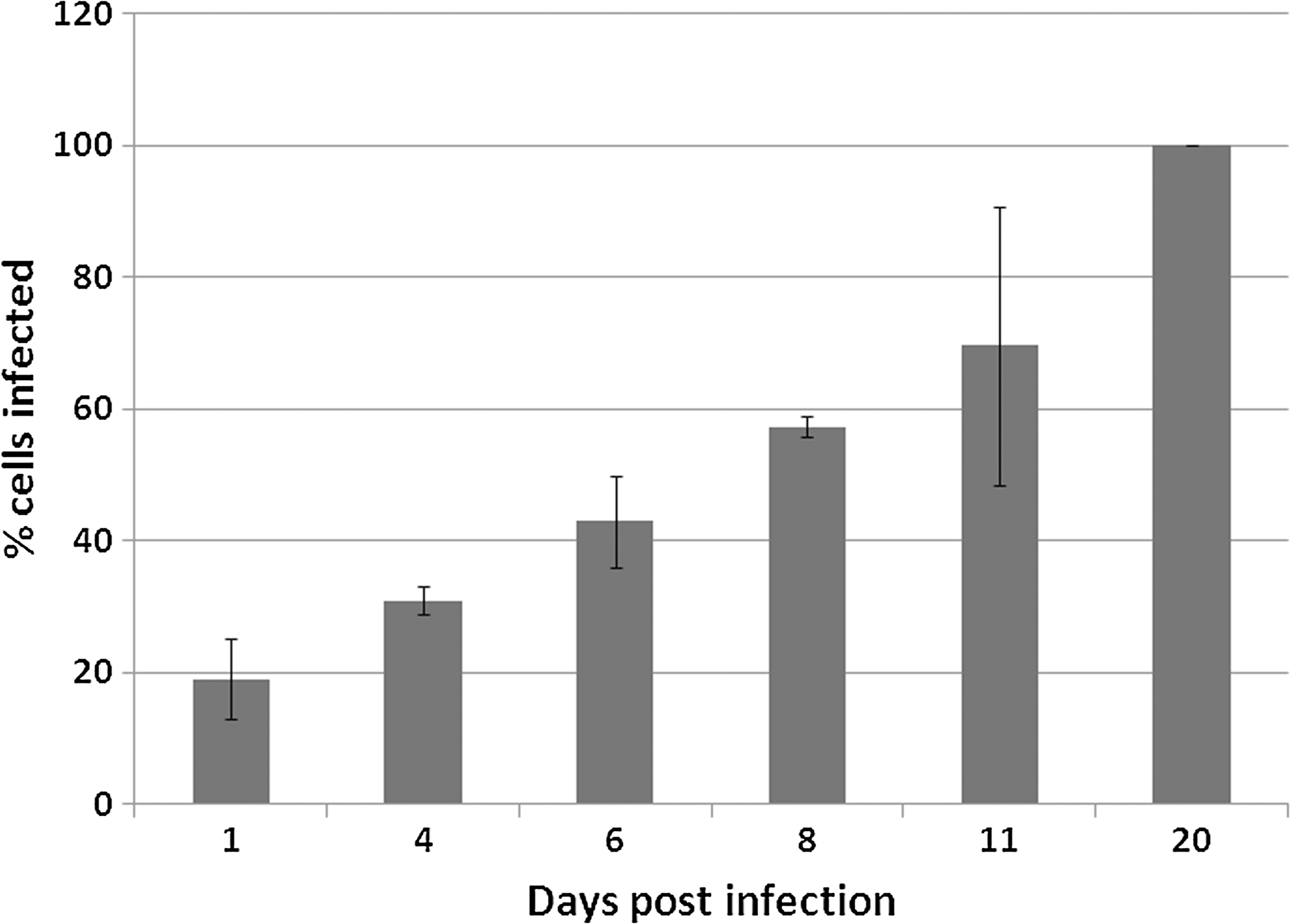
Average percentage of D. melanogaster S2 cells containing at least one R. felis LSU bacterium at selected time points. Results are the averages of three separate time-course infection experiments (mean±standard deviation [SD]).
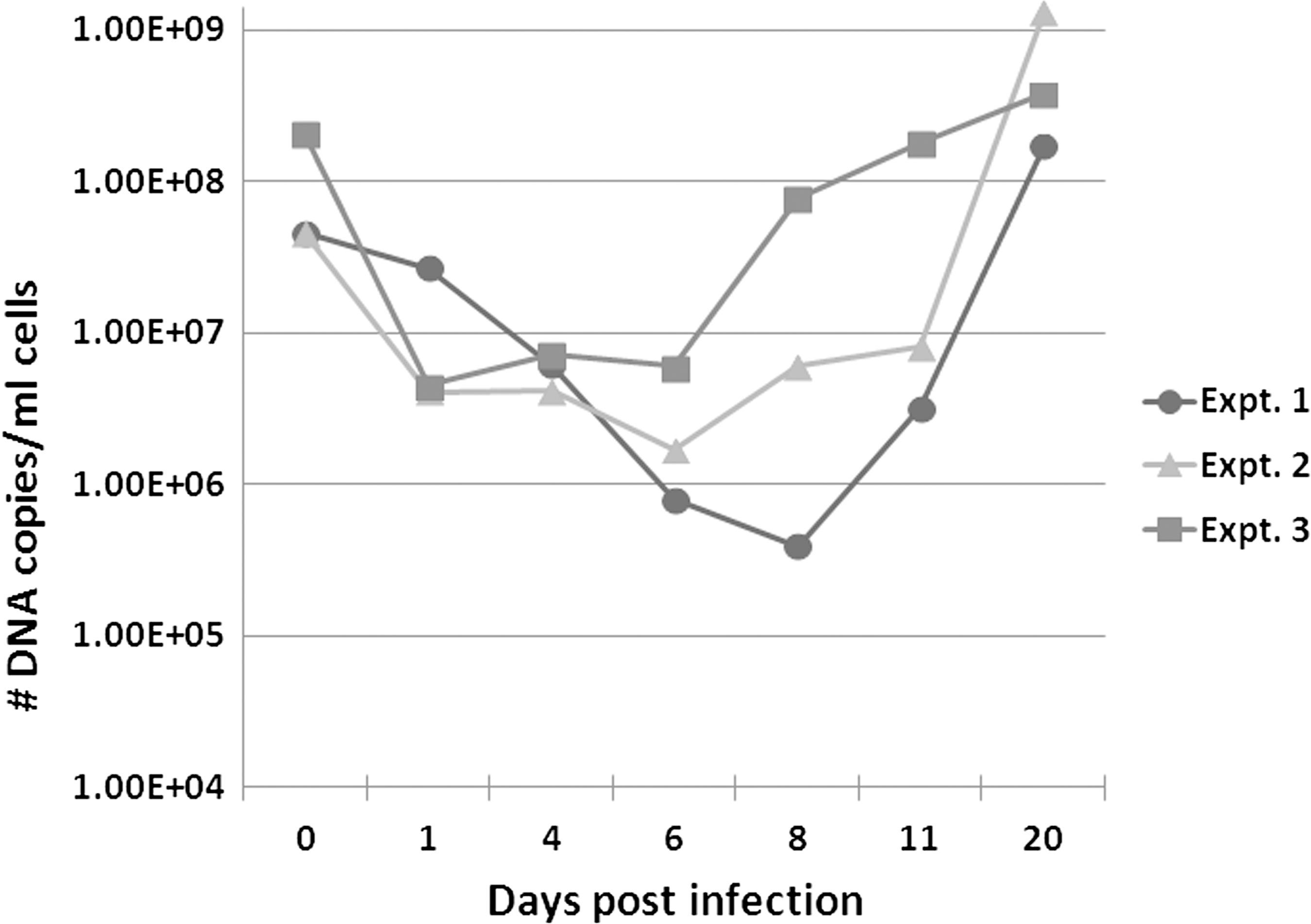
Growth of R. felis LSU in D. melanogaster S2 cells. Three independent time-course infection experiments in S2 cells are shown. The number of DNA copies present at the indicated time points was quantitated using a R. felis–specific qPCR assay. No rickettsial copies were detected in uninfected/negative control cells (data not shown).
The number of DNA copies of R. felis were also compared at selected time points in the infected S2 cells. Approximately 1×108 DNA copies were used to initiate infections in the S2 cells for each of the three independent time-course experiments; this is indicated by 0 dpi in Figure 2. Following the initial infection, a gradual decrease was observed in the number of R. felis DNA copies from 1 to 6 dpi (Fig. 2). After 6 dpi, a subsequent increase in the number of DNA copies present was observed, and by 20 dpi a greater number of DNA copies were always observed compared to the number present at 0 dpi (Fig. 2). For example, in time-course experiment 1, a total of 4.6×107 DNA copies were used to initiate infection (0 dpi) and by 20 dpi a total of 1.7×108 copies/mL of cells were detected by qPCR. In experiment 2, a total of 4.6×107 DNA copies were used for infection (0 dpi) and by 20 dpi a total of 1.3×109 copies/mL of cells were detected by qPCR. In experiment 3, a total of 2.1×108 DNA copies were used for infection (0 dpi), and by 20 dpi a total of 3.8×108 copies/mL of cells were detected by qPCR (Fig. 2). As observed in Figure 2, the greatest increase in the number of DNA copies could usually be observed between 11 and 20 dpi for each of the time-course experiments.
In addition to scoring cells for the presence/absence of rickettsial organisms and quantitating the number of DNA copies at selected time points, the S2 cells used for the time-course experiments were also visualized using Diff-Quik and Acridine Orange staining. Diff-Quik and Acridine Orange staining allowed for a visual observation of R. felis in the S2 cultures from 2 to 20 dpi (Figs. 3 and 4). As seen by Diff-Quik stain (Fig. 3), a large number of rickettsiae could be observed outside the cells during early time points, such as 2 and 6 dpi. At 9 dpi, cells heavily infected with R. felis were observed with Diff-Quik stain; and by 12 and 20 dpi, rickettsiae were observed to heavily parasitize cells as well as be present in the extracellular space(s) (Fig. 3). Although rickettsiae were often observed in the extracellular spaces during the early time points of infection, it was also possible to always find cells that were very heavily infected with R. felis during the early time points. This was evidenced by Acridine Orange staining at time points 4, 6, and 8 dpi (Fig. 4) and Diff-Quik staining at 6 dpi (Fig. 3). Additionally, R. felis could regularly be observed occurring in long chain-like formations, often between 11 and 20 dpi (Figs. 3 and 4). Staining R. felis–infected cells with Diff-Quik and Acridine Orange stains also allowed for the visualization of cytopathic changes in the S2 cells following infection with R. felis. No immediate cytopathic changes were observed in the S2 cells following infection with R. felis. At approximately 11–12 dpi, a subset of cells began to appear more heavily vacuolated; by 20 dpi, many of the cells were lysed, and free rickettsiae appeared throughout the extracellular space(s). The intact cells present at 20 dpi were always very heavily parasitized by R. felis (Figs. 3 and 4).

Diff-Quik staining of R. felis in the cytoplasm and extracellular space(s) of D. melanogaster S2 cells at various time points, as indicated by arrows. No rickettsiae were observed in uninfected cells (negative). Images were captured using an Olympus BX43 microscope and DP72 camera with a 10× ocular and 100× objective.
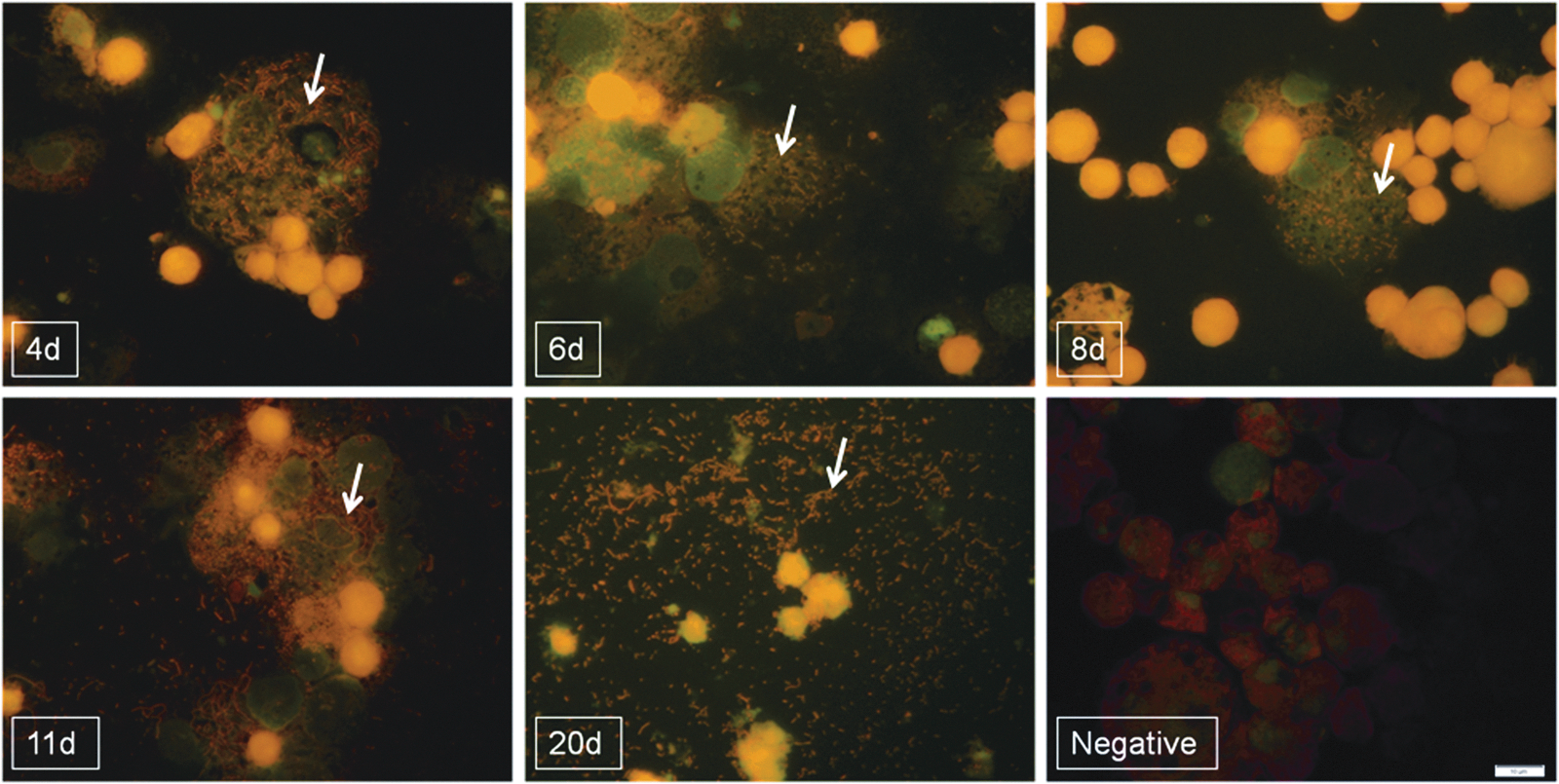
Acridine Orange staining of R. felis LSU in D. melanogaster S2 cells at various time points, as indicated by arrows. No rickettsiae were observed in uninfected cells (negative). Images were captured using an Olympus BX43 microscope and DP72 camera with a 10× ocular and 100× objective.
TEM was also used to better visualize the biological characteristics of R. felis within the S2 cells. At 19 dpi, S2 cells heavily infected with R. felis were observed by TEM. Rickettsiae of elongated and round forms could be observed freely within the cytoplasm of the cell (Fig. 5). The longer rickettsiae were approximately 1–1.8 μm in length and 0.3 μm in width/diameter; the round rickettsiae were approximately 0.3–0.4 μm in diameter (Fig. 5). Electron-lucent “halos,” presumably the rickettsial slime layers, could also be observed surrounding the R. felis found within the cytoplasm of the cells (Fig. 5). A thick membrane could be discerned surrounding the rickettsiae found in the cytoplasm of the cells. Additional TEM analysis will be necessary to observe the periplasmic space associated with typical rickettsial membranes, as seen in previous studies involving the in vitro growth of R. felis (Sunyakumthorn et al. 2008).
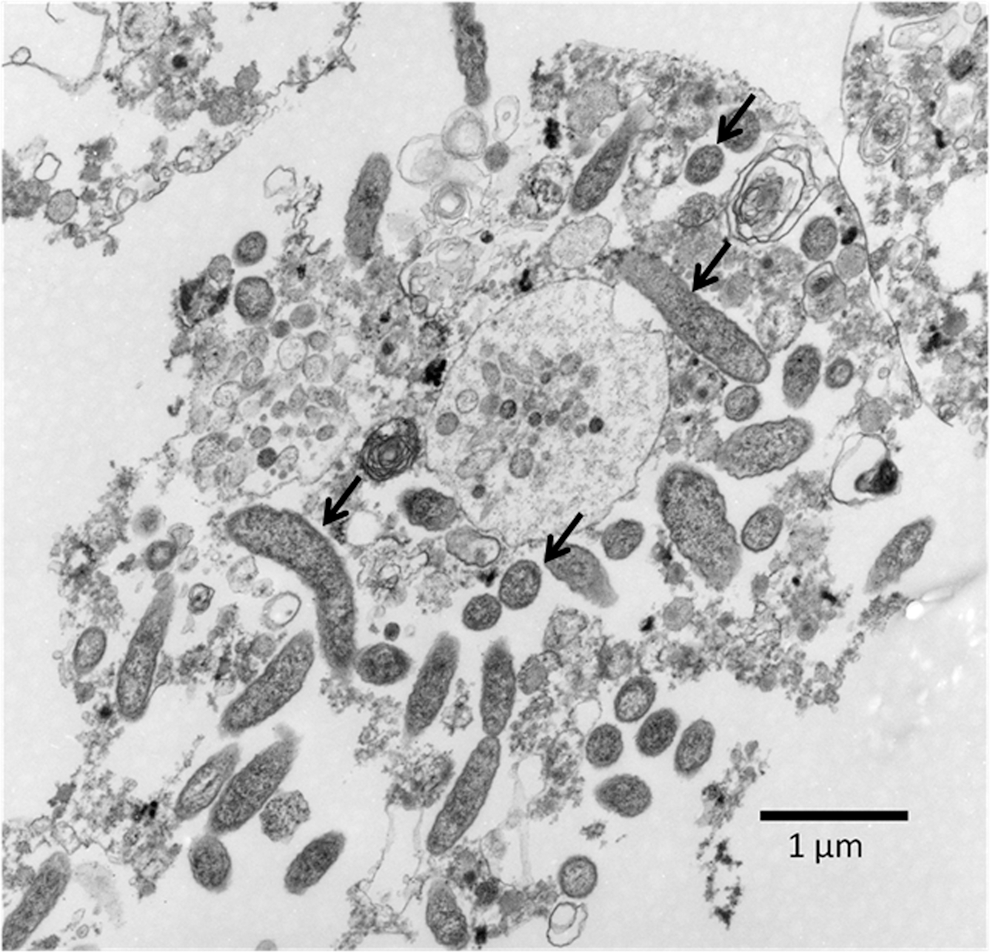
Transmission electron micrograph of R. felis LSU in D. melanogaster S2 cell at 19 days postinfection (dpi); arrows indicate examples of round and elongated rickettsiae. No rickettsiae were observed in uninfected cells (data not shown).
Consequently, R. felis can be propagated in Drosophila S2 cells and appears to complete its replication cycle as evidenced by molecular quantitation and cytological staining techniques. To date, we have passaged R. felis in the S2 cells for upward of 20 passages, which has included the use of frozen seed stocks (−80°C) to initiate infections in the S2 cells.
R. felis grown in S2 cells is infectious to other cells
To determine if R. felis strain LSU was maintaining its infectivity when grown in Drosophila S2 cells, we used R. felis that had been grown in S2 cells for 10–15 passages to infect C6/36 and/or Vero cells. Both of these cell lines have been used in the past for the propagation of R. felis (Horta et al. 2006, Saisongkorh et al. 2011). Time-course infections were simultaneously conducted in S2, C6/36, and Vero cells, and at selected time points DNA extractions and Diff-Quik and/or Acridine Orange staining were performed on infected cells. When the number of DNA copies at selected time points was compared between S2 cells and C6/36 cells, a similar trend was observed in two independent experiments (Fig. 6). From the initiation of infection (0 dpi) until approximately 6 dpi, we observed a decrease in the number of DNA copies in both the S2 and C6/36 cells (Fig. 6). After 6 dpi, a subsequent increase in the number of DNA copies was observed at each time point in both the infected S2 and C6/36 cells (Fig. 6). Moreover, a greater number of R. felis DNA copies were present at 20 dpi in the S2 and C6/36 than were used to initiate infection (0 dpi) (Fig. 6). During the first time-course infection, 8.4×107 DNA copies were used to initiate infection (0 dpi), and by 20 dpi 5.9×108 DNA copies were detected in the S2 cells and 1.4×108 copies were detected in the C6/36 cells (Fig. 6). Similar results were observed in the second time-course infection: 1.4×108 copies were used to initiate infection (0 dpi) and at 20 dpi, 3.3×108 DNA copies were detected in the S2 cells, and 1.9×109 copies were detected in the C6/36 cells (Fig. 6). The overall average largest increase in the number of rickettsial DNA copies occurred from 15–20 dpi in both the S2 and C6/36 cell lines (Fig. 6).
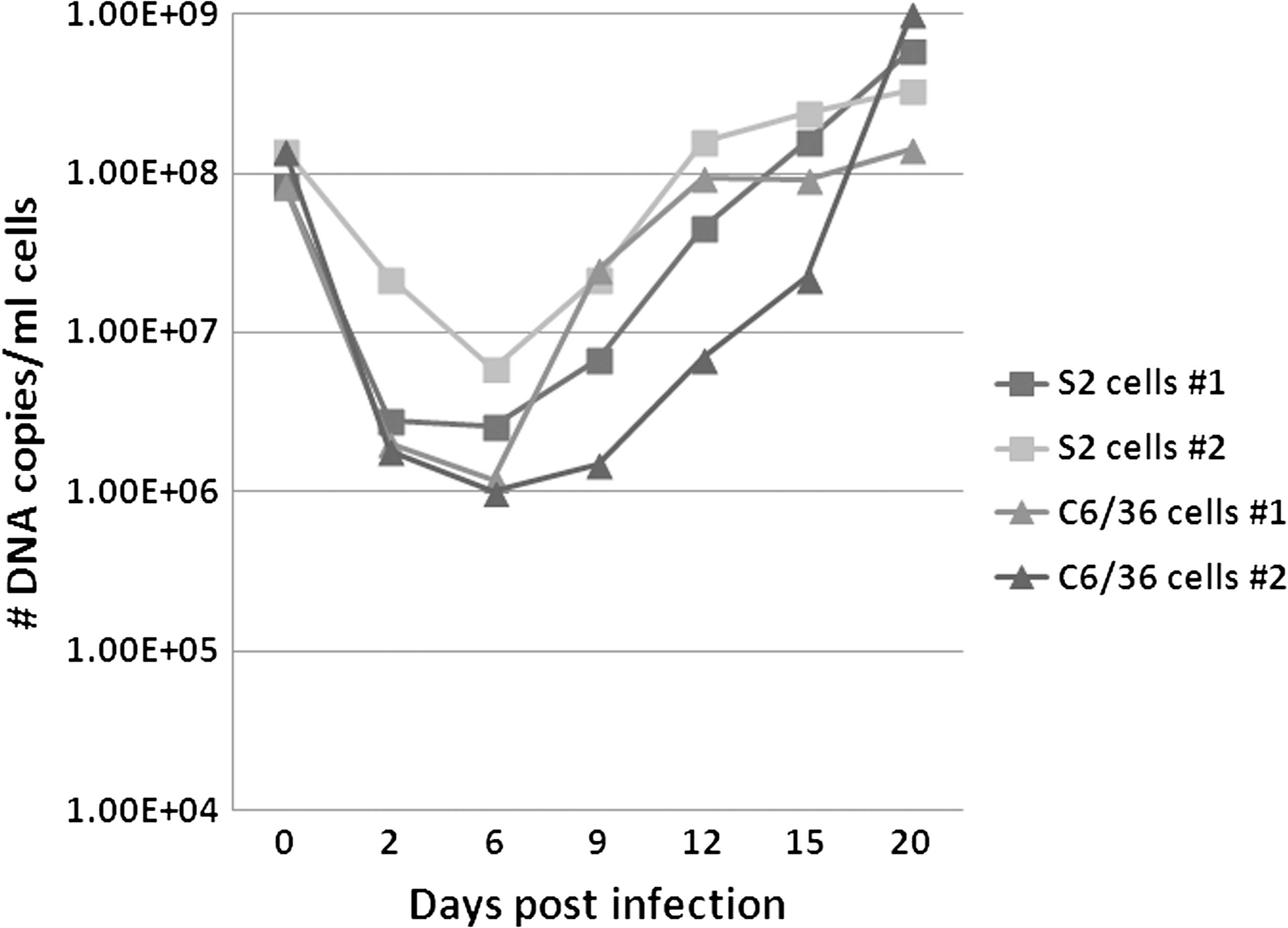
Comparison of growth of R. felis LSU in D. melanogaster S2 cells and mosquito C6/36 cells. Two simultaneous, independent time-course infection experiments are shown. The number of DNA copies present at the indicated time points was quantitated using a R. felis–specific qPCR assay. No rickettsial copies were detected in uninfected/negative control cells (data not shown).
In contrast to the S2 and C6/36 cell lines, we observed little to no increase in the number of R. felis DNA copies throughout the time course infection in the Vero cell line (Fig. 7). In both the S2 and Vero cells, an initial decrease in the number of DNA copies was observed from the initiation of infection (0 dpi) until 6 dpi (Fig. 7). Although a subsequent increase in the number of R. felis DNA copies could be observed at each time point from 6 through 20 dpi in the S2 cells, the overall number of DNA copies detected in the Vero cells remained relatively stagnant beyond 6 dpi, with only small, inconsistent increases in the number of copies between 9–12, 12–15, and 15–20 dpi (Fig. 7). Therefore, an overall loss in the number of DNA copies present in the Vero cells was observed from the initiation of infection (0 dpi) to 20 dpi (Fig. 7). This is in direct contrast to the results observed in the S2 and C6/36 cells (Fig. 7).
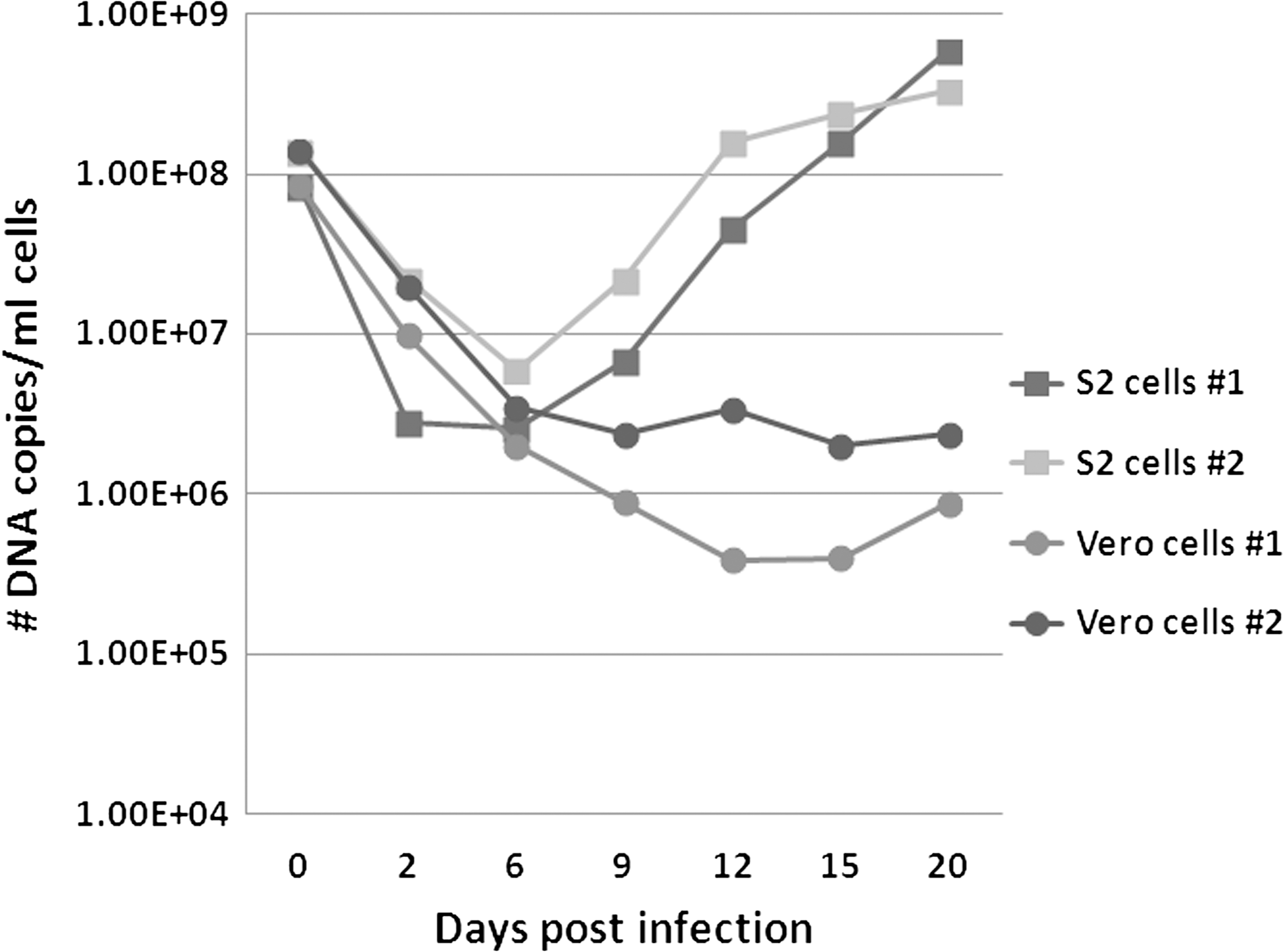
Comparison of growth of R. felis in S2 cells and Vero cells. Two simultaneous, independent time-course infection experiments are shown. The number of DNA copies present at the indicated time points was quantitated using our R. felis qPCR assay. No rickettsial copies were detected in uninfected/negative control cells (data not shown).
Diff-Quik staining was also used to visualize the presence of R. felis within the C6/36 and Vero cells throughout the time-course infections. We observed similar dynamics of R. felis infection in the C6/36 cells as compared to the S2 cells. At 2 dpi, the majority of R. felis organisms could be observed in the extracellular spaces of the C6/36 cells; by 6 and 9 dpi, a large number of cells could be found to be parasitized by R. felis; and by 12 and 20 dpi, a large number of cells were heavily infected with R. felis, lysed cells were observed, and many organisms could be observed in the extracellular space(s) (data not shown). Compared to the S2 cells, we did not observe the frequent formation of chain-like groups of R. felis in the C6/36 cells.
The opposite growth dynamics of R. felis LSU in S2 and C6/36 cell lines were observed in the Vero cells when visualized with Diff-Quik stain. While we were able to observe a few R. felis organisms in the extracellular space(s) of the Vero cells at early time points (2 and 6 dpi), little to no rickettsiae were observed at time points of 9 dpi and beyond (data not shown). No R. felis were observed in uninfected cells in all experiments.
To further visualize the presence of R. felis within the S2, C6/36, and Vero cells, we used a polyclonal antibody directed against spotted fever group rickettsiae. Just as observed with Dif-Quik staining, a large number of R. felis organisms could be observed extracellularly in all three cell lines at 2 dpi (Fig. 8). At 9 dpi, R. felis could be observed intracellularly in both the S2 and C6/36 cells (Fig. 8). And by 20 dpi, R. felis could be observed in large numbers both intra- and extracellularly in the S2 and C6/36 cells (Fig. 8). In the Vero cells, R. felis was detected at all time points extracellularly, rarely intracellularly, and few to no rickettsiae were apparent at 20 dpi in comparison to the S2 and C6/36 cell lines (Fig. 8).
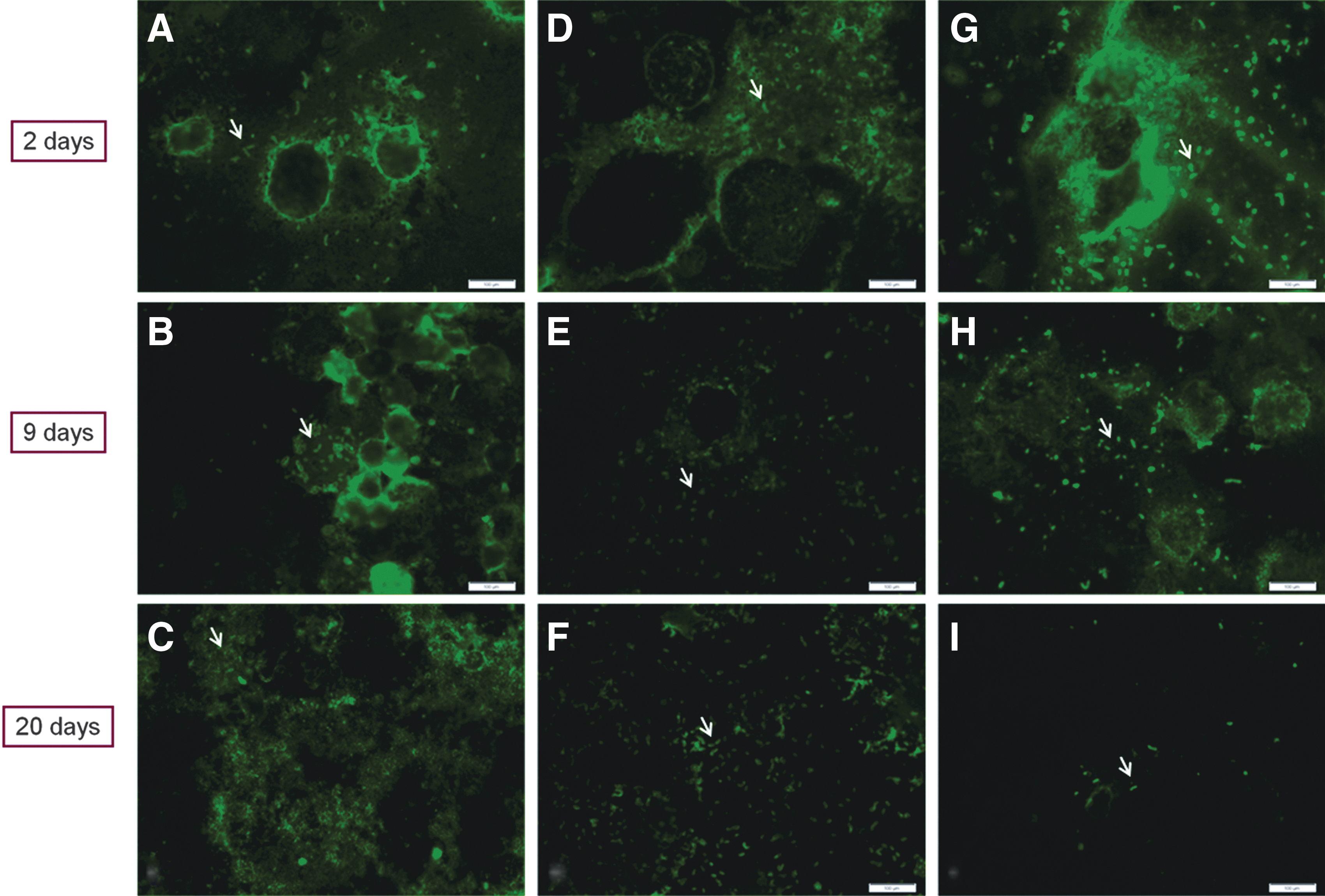
Immunocytochemical detection of R. felis in S2, C6/36, and Vero cells at various time points, as indicated by arrows. Rickettsiae were detected by incubation of the cells with polyclonal spotted fever group primary antibody and goat anti-rabbit immunoglobulin G (IgG) secondary antibody in infected cells as seen in infected S2 cells (
Discussion
The data presented here reveal that R. felis strain LSU can be cultured and maintained in D. melanogaster S2 cells. Infection of the S2 cells was monitored using molecular and cytological methods. Observations of initial decreases in the number of R. felis DNA copies were observed in the S2 and C6/36 cells, followed by subsequent increases in the number of DNA copies. It is possible that this observation may represent a lag phase or adaptation to the host cell followed by an exponential growth phase of R. felis. A lag phase of 2–3 days has been described for Rickettsia helvetica cultivated in Vero cells, 12 h for Ehrlichia ruminantium cultivated in bovine endothelial cells, and 0–7.5 h for Rickettsia prowazekii cultured in chicken embryo cells (Wisseman et al. 1976, Marcelino et al. 2005, Elfving et al. 2012). Moreover, the length of the lag phase was dependent upon the bacterial growth phase and/or the morphological form from which the infecting seeds were isolated (Wisseman et al. 1976, Marcelino et al. 2005). Thus, it could be hypothesized that the lag phase of 6–8 days observed in our studies may be the result of the replicative state of our R. felis, the bacterial growth phase from which they were isolated for use in our infection experiments, and/or the necessity of medium conditioning for enhanced growth. More experiments will need to be performed to confirm this hypothesis.
The greatest number of DNA copies observed across the time course infection experiments always occurred at 20 dpi in both the S2 and C6/36 cells. In previous studies using mammalian culture systems, maximum DNA copy numbers have been reported to occur at 6–7 dpi for R. helvetica (Elfving et al. 2012), 4 dpi for R. slovaca (Boldis and Spitalska 2010), and 2–3 dpi for R. rickettsii (Eremeeva et al. 2003). Interestingly, R. slovaca growth in ticks was reported to reach maximum DNA copy number at 12 dpi in Ixodes ricinus and at 21 dpi in Dermacentor marginatus ticks (Boldis and Spitalska 2010). Thereby, the maximum DNA copy number at 20 dpi infection that we observed in the S2 cells may possibly more closely mimic the growth observed in an arthropod vector than that which is observed using mammalian culture systems. These types of observations reveal the attractiveness of using S2 cells as an alternate culture system for the study of rickettsial agents.
Our TEM images revealed that both a rod shape and rounder form of R. felis LSU appeared to be present in the S2 cells at 19 dpi. Both forms were of normal size for rickettsial pathogens (0.3–0.5 μm by 0.8–2.0 μm), and the round form(s) most likely represents cross-sections of the elongated form(s). Sunyakumthorn et al. (2008) observed the presence of long form (up to six times the normal rickettsial length) R. felis when the bacteria were cultured in the tick cell line ISE6. The polymorphism of the R. felis forms in the ISE6 cultures was influenced by rickettsial density, nutrient availability, and the length of time maintained in cell-free conditions (Sunyakumthorn et al. 2008). Similarly, longer forms of R. bellii (from 3 μm to 15 μm) have also been reported depending upon nutrient availability of the cell culture (Philip et al. 1983, Labruna et al. 2004). Although we did not observe these long forms in our S2 cultures, further experiments manipulating incubation times, media formulations, and cell-free culture using the S2 cells could help to determine if the formation of long-form R. felis is exclusive to the tick system or can be induced in other arthropod culture systems.
Cytopathic effects were minimal at early time points in the infected S2 cells (and in the C6/36 and Vero cells), with the most noticeable vacuolation of cells occurring between 11 and 12 dpi. A lack of marked cytopathic effects was also observed when R. felis was grown in tick cells (Pornwiroon et al. 2006, Sunyakumthorn et al. 2008). When R. felis LSU was grown in mosquito Sua5B cells, clumping and lysing of the cells was reported (Sakamoto and Azad 2007). Thus, limited cytopathic effects observed in the infected S2 cells were not drastically different from reports using other arthropod cells lines.
In conclusion, we have demonstrated that R. felis LSU can be successfully propagated and maintained in S2 cells. In addition, we have recently used the S2 cells line to grow and isolate R. felis from fleas (C. felis) associated with an outbreak of febrile illness in humans, in Orange County, California (Luce-Fedrow and Richards, unpublished data). To date, no human isolates of R. felis have successfully been grown in vitro. S2 cells may possibly provide the solution to this obstacle in the future due to the ease of both culturing and growing freshly isolated R. felis in S2 cells.
Furthermore, much remains to be learned concerning the establishment and transmission of R. felis in and between its arthropod and mammalian hosts. Although other arthropod cell lines, like C6/36 and ISE6, have been previously used to grow R. felis, the Drosophila cell line system represents a powerful tool that has the potential for providing answers to questions surrounding R. felis host–pathogen interactions using molecular genetics techniques. Most recently, the Drosophila system has been used to identify key genes that are upregulated and contribute to establishing infections of E. chaffeensis, a rickettsial order member, both in vitro and in vivo in D. melanogaster (Von Ohlen et al. 2012). A human homolog of one of the genes identified using the Drosophila system was then tested/targeted with double-stranded (ds) RNA-induced silencing and found to have an impact on the establishment of E. chaffeensis infections in vitro (Von Ohlen et al. 2012). This type of study demonstrates the usefulness that the Drosophila system may provide for discerning key host–pathogen components particular to rickettsial infections. Consequently, the use of Drosophila for studying rickettsial pathogens is advantageous due to the ease of maintenance of both S2 cells and D. melanogaster, the ability to grow both insect and tick-borne rickettsiae in S2 cells, and the vast array of molecular tools that are available when using Drosophila for studying host–pathogen relationships.
Footnotes
Acknowledgments
This work is supported by the Global Emerging Infections Surveillance and Response System, a Division of the Armed Forces Health Surveillance Center; work unit number 0000188M.0931.001.A0074.
Author Disclosure Statement
There is no conflict of interest for all authors.
The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Department of the Navy, Department of Defense, nor the US Government.
A.L.R. is an employee of the US Government, and this work was prepared as part of his official duties. Title 17 U.S.C. §105 provides that “Copyright protection under this title is not available for any work of the United States Government.” Title 17 U.S.C. §101 defines a US Government work as a work prepared by a military service member or employee of the US Government as part of that person's official duties.