
Abstract
The complete nucleotide sequences of two West Nile virus (WNV) strains isolated in Argentina were determined. Phylogenetic trees were constructed from the aligned nucleic acid sequences of these two strains along with other previously published complete WNV genome sequences. Phylogenetic data showed that both strains belonged to clade 1a of lineage 1 and clustered in a subclade with American strains isolated during 1999–2002. These results suggest two independent routes of introduction of WNV in Argentina and that the virus could have been circulating in Argentina for some time before being isolated.
Introduction
W
The genome of WNV is approximately 11,000 nucleotides in length, depending on the strain, with three structural genes (capsid [C], membrane [prM-M], and envelope [E]) and seven nonstructural genes (NS1, NS2a, NS2b, NS3, NS4a, NS4b, and NS5) (Granwehr et al. 2004). Currently, WNV is divided into five major lineages (Bondre et al. 2007). WNVs from lineages 3 and 4 have not been isolated from humans and currently have no known association with epidemics or epizootics. Lineage 5 viruses are associated with epidemics and epizootics in India and were formerly grouped within lineage 1; however, recent data support their classification as a unique lineage (Bondre et al. 2007). WNV lineage 2 strains have been isolated from sub-Saharan Africa, Madagascar, and Eastern Europe countries. Although WNV lineage 2 originally was not associated with central nervous system compromise (Lanciotti et al. 2002), recently, neurological disease has been reported both in horses and humans (Venter et al. 2009, 2010, Barzon et al. 2013a, b).
Lineage 1 strains are found in North America, Europe, Africa, Asia, and Australia and are frequently associated with epidemics and epizootics in which central nervous system disease and fatalities are prominent features. Lineage 1 viruses are grouped into three clades: Clade 1A contains isolates from Africa, Europe, the Americas, and Israel. The Australian Kunjin viruses belong to clade 1B exclusively found in Australia, whereas isolates from India form clade 1C. Clade 1A isolates are grouped in six clusters: Cluster 1 includes isolates from Egypt, Ethiopia, India, and Europe; cluster 2 has been implicated in most European and northern African epidemics over the last 20 years; cluster 3 is comprised of isolates collected in Astrakhan between 1999 and 2005; cluster 4 contains all isolates from the Americas, in addition to a single isolate each from Israel in 1998, Tunisia in 1997, and Hungary in 2003. The recently described clusters 5 and 6 include two isolates from West Africa (Senegal isolated in 1979 and Nigeria isolated in 1965) and an isolate from Central Africa Republic in 1967 (May et al. 2010), respectively.
The purpose of this study was to determine the relationships between the two WNV strains isolated in Argentina in 2006 and other previously described WNV strains. The complete genome nucleotide sequences of two WNV strains recently isolated in Argentina were determined and the phylogenetic relationships analyzed.
Materials and Methods
The isolation history of both WNV strains ArEq001 and ArEq003 isolated in Argentina from the brains of horses that died from encephalitis in February and March of 2006 have been published elsewhere (Morales et al. 2006). RNA was extracted from 140 μL of cell culture supernatant of the original isolations made in African green monkey (Vero) cell culture with the QIAmp Viral RNA Kit (Qiagen) according to the manufacturer's protocol. DNA templates for sequencing were generated as follows: The open reading frame (ORF) regions of WNV of ArEq001 and ArEq003 were amplified by nested RT-PCR. Twenty and 43 WNV-specific primer pairs were used in the first and second rounds of amplification, respectively. Fragments sized from 200 to 1100 bp were generated and analyzed on a 2% agarose gel. DNA bands of the correct size were excised and purified with the QIAquick Kit (Qiagen) according to the manufacturer's protocol. Both strands of the purified DNAs were sequenced with the use of a Big Dye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) and a total of 77 WNV-specific primers. Nested RT-PCR amplification and nucleic acid sequencing were performed as previously described (Lanciotti et al. 1999), and a complete list of the sequencing primers used is available upon request.
The nucleotide and deduced amino acid sequences derived from each of both WNV Argentine strains were aligned with one another and with 48 previously published WNV strains obtained from the GenBank (strains from lineages 1) (Table 1). Sequences were edited and aligned using the BioEdit program and the ClustalW method (available from
Results
In a previous study, we described the detection of WNV RNA from three equines in Argentina during the summer of 2006 (Morales et al. 2006). This work included a preliminary phylogenetic analysis of viral nucleic acid sequence data, on the basis of two short (356 bp and 313 bp) fragments of the C/preM and NS5 coding regions, showing the inclusion of all three viral strains in WNV linage 1. In the present report, we extended the genetic characterization of two of these viral strains, one from each of the locations where the strains were isolated. The complete genome sequences of ArEq001 and ArEq003 were derived, with the exception of the terminal amplification primer sequences—the first 25 bases of the 5′ end and the terminal 24 bases of the 3′ end.
Pairwise alignments of ArEq001 and ArEq003 with 48 other WNV complete genome sequences (Table 1) revealed that the two Argentine viruses each shared the highest nucleotide identity with the WNV NY1999 strain—99.5% identity (50 differences) between ArEq001 and NY1999, and 99.3% (71 differences) between ArEq003 and NY1999 (Table 2). Table 2 displays the nucleotide and amino acid identities for a subset of six WNV strains analyzed. A complete nucleotide genome BLAST analysis of the two Argentine viruses confirmed that each virus was most closely related to WNV NY1999 (data not shown). Interestingly, ArEq001 and ArEq003 demonstrated a greater degree of difference with respect to one another than each to the NY1999 virus—99.0% nucleotide identity (111 differences) between ArEq001 and ArEq003 (Table 2). Fourteen amino acid differences were observed between the two Argentine strains; six of them are conservative: V→I (cap-113), I→V (preM-14), A→V (NS1-171), E→D (NS2a-3), G→A (NS4a-88), V→A (NS4b-23), and eight are nonconservative: G→R (NS2a-97), H→Y (NS2a-118), M→I (NS3-213), F→L (NS4b-34), A→T (NS4b-116), Q→P (NS5-431), H→Y (NS5-577), and D→N (NS5-791). A comparison of the amino acid substitutions between the two Argentine strains and other nine WNV published full-length amino acid sequences is displayed in Table 3. In ArEq001 we observed five amino acids not present in any other strain (NS1-171, NS2a-3, NS2a-97, NS3-213, and NS5-431). ArEq003 presented only two amino acids not shared with any other strain (NS4a-88 and NS5-577).
Amino acid percent identities are shown in the upper right quadrant, and nucleotide identities are shown in the lower left quadrant.
Volgograd 2000 human, AY278442; Kenya 1998, AY262283; Caucasus 1988, A Y277251; Australia Kunjin 1960, D00246; India 1980 human, DQ256376; Uganda 1937 human B956, AF394221; Rabensburg 1997, AY365264.
Phylogenetic trees were constructed from the aligned nucleic acid data by using the complete genome of Argentine strains and 48 available sequences of WNV from lineages 1A obtained from GenBank (Table 1). Phylogenetic trees constructed using distance methods (neighbor joining), tree-searching methods (maximum parsimony), and Bayesian analysis generated trees with the same overall topology. Figure 1 displays the Bayesian tree. WNVs worldwide were grouped into five major lineages, as previously published (Bondre et al. 2007). The two WNVs from Argentina were placed within lineage 1 in the NY1999/Israel1998 clade (clade 1A-4). Within this clade the viruses ArEq001 and ArEq003 are grouped with several WNVs from the northeastern United States isolated in 1999 and 2000; the NY1999 genotype was described by Davis et al. (2005). An analysis of phlyogenetically informative sites reveals that the ArEq003 virus and the Israel1998 virus share identical nucleotides at four sites (315, 3984, 3990, and 3993) that differ from all other WNVs, further suggesting a close relationship of these two viruses with one another and a link to a common ancestor to all Western Hemisphere WNVs.
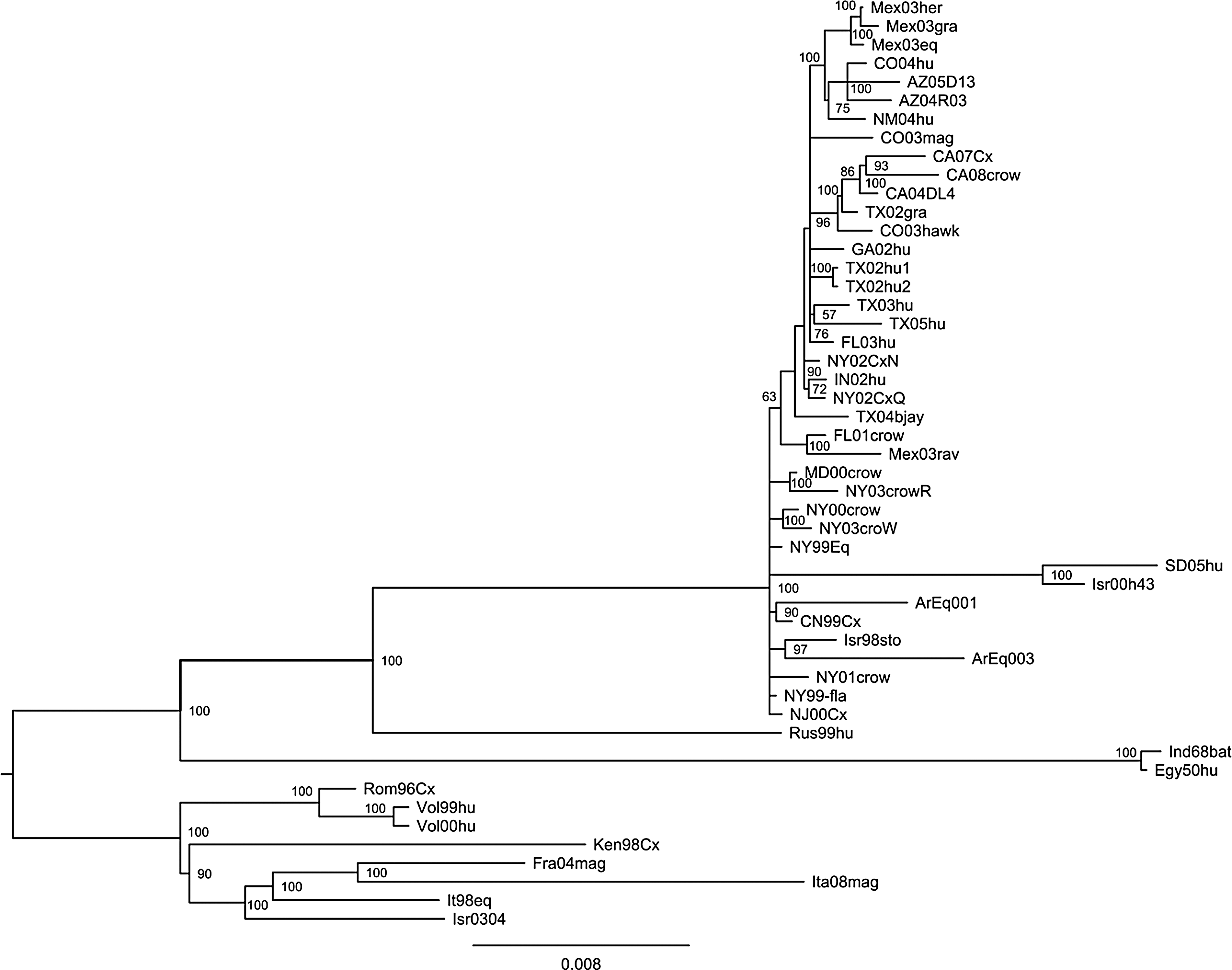
Phylogenetic tree constructed by utilizing the complete genome sequence of West Nile virus (WNV) strains from lineage 1 (abbreviations are listed in Table 1). Bayesian analysis was performed using MrBayes 3.1.2 (Ronquist and Huelsenbeck 2003). jModelTest was used to select the best-fitting model, proving to be TIM+G+I Two parallel runs of four (three heated) Metropolis-coupled Markov chain Monte Carlo algorithms were run for 10,000,000 iterations, with subsampling every 2000 samples.
Discussion
In the summer of 1999, WNV was introduced into North America and has spread rapidly throughout all the United States and Canadian provinces, as well as Mexico, the Caribbean islands, and Colombia over the next 3 years. WNV infections first appeared in human residents of the Cayman Islands and the Florida Keys in 2001, and in apparently healthy Jamaican birds sampled early in 2002. Serologic evidence of WNV infection in 2002 was detected in horses, chickens, and resident free-ranging birds in Guadeloupe, the Dominican Republic, and eastern Mexico. In 2003, WNV was found in Mexico and northern Central America, and serologic evidence was detected in the Bahamas, Puerto Rico, and Cuba (Puppo et al, 2006).
In 2004, the first serologic evidence of WNV activity in South American ecosystems surfaced in Colombia and Trinidad, where domestic animal WNV-neutralizing antibodies were detected (Castillo-Olivares and Wood 2004, Davis et al. 2005, Komar and Clark 2006, Artsob el al. 2009). There have been recent reports of serological evidence in horses in Venezuela (2006) and in Brazil (2009) (Bosch et al. 2007, Pauvolid-Correâ el al. 2011). The first known autochthonous human cases of WNV infection in South America occurred during 2005 in Colombia, followed by the first known human WNV encephalitis cases in 2006 with additional human cases recorded in 2007 (Mattar et al. 2005, Artsob et al. 2009). In a previous study, we reported the isolation of three WNV strains from brains of dead horses in two different stud farms, 200 km away from each other, in Argentina during the summer of 2006 (Morales et al. 2006).
In the present report, we extend the genetic characterization of two of the three equine WNV strains mentioned above to obtain full polyprotein sequences. One strain from each farm (ArEq001 and ArEq003) was sequenced. The sequence data present two interesting and puzzling observations. First is the high degree of nucleotide dissimilarity between the two strains ArEq001 and ArEq003 (111 nucleotide differences and 14 amino acid differences) isolated in Argentina during the same year within 200 km of one another. This degree of difference is far greater than expected based upon the extent of evolution observed among North American WNVs separated by much greater distance and time. Analysis of WNV strains in North America from 1999 to 2008 reveals an evolutionary rate of approximately seven to eight nucleotide changes per year per genome. After 9 years of movement into diverse ecological zones and evolution in North America, the two most distantly related viruses, the CA 2008 crow strain and the Mexico 2003 raven strain, differ by 83 nucleotides. A difference of 111 nucleotides between the two Argentine viruses suggests either two separate introductions into Argentina of two distinct WNV strains or a single WNV strain introduction with divergent evolution for over 10 years, assuming a rate of evolution similar to that observed in North America.
Of note is the detection of neutralizing antibodies to WNV detected in 2005 in horses from different places in Argentina (M.A. Morales 2005, unpublished data), confirming that WNV was circulating in Argentina at least 1 year before being isolated. Additional evidence of WNV circulation in Argentina was provided by serologic detection of WNV antibodies in birds between 2005 and 2006 in Chaco, Córdoba, Tucumán, and Entre Rios Provinces (Diaz et al. 2008). After the detection of WNV in equines and birds, we detected the first human WNV encephalitis cases in Argentina in 2006 with additional human cases being recorded in 2007 and 2010 (Artsob et al. 2009). Second, the nucleotide sequence data and the resulting phylogenetic trees strongly suggest that ArEq003 is not a progeny virus of North American WNVs, but rather the result of an introduction of a WNV strain from an undefined location that shares a common ancestor with North American WNVs.
Phylogenetic trees that include WNVs isolated from Mexico (2003–2004), Puerto Rico (2006), and Guatemala (2007) all demonstrate a close relationship of these viruses with US WNVs. The Mexican, Puerto Rican, and Guatemalan WNVs are always placed in branches of the tree along with US WNVs, indicating that these viruses in Latin America originated from the United States (Deardorff ER 2006, unpublished data). The phylogenetic trees constructed by both distance and tree searching methods consistently place ArEq003 in a separate branch with the Israel1998 WNV and separated from the North American WNV strains, indicating that ArEq003 did not originate by southward movement from North America, but rather from a common ancestor with the Israel1998 WNV. The percent nucleotide identity data in Table 2 support this concept of an independent introduction; the ArEq003 strain shares the highest percentage identity with NY1999 and the dissimilarity increases between ArEq003 and WNV strains isolated in each subsequent year North America.
In 2005, Davis et al. showed evidence for the emergence of a dominant genotype in North America (WN02) characterized by three signature nucleotide mutations in the prM and E protein genes prM 660 (C→U), E1442 (U→C) and E 2466 (CU) (Davis et al. 2005, 2007). Argentine strains do not posses any of these mutations, confirming that ArEq001 did not follow the evolutionary migration pattern of other WNV strains found in Mexico that possess these mutations. Finally, the existence of four signature sites shared by ArEq003 and the Israel1998 virus further supports the notion that Israel1998 and ArEq003 share a common ancestor that was the source of the introductions of WNV into New York in 1999 and into Argentina at some undefined time. The phylogenetic trees and the pairwise nucleotide data are inconclusive concerning the origins of ArEq001; however, the number of amino acids unique to this strain (5, described above) suggest that this virus may also have originated from somewhere other than North America.
The data presented here provide the first indication that WNV may have moved into the Western Hemisphere on more than one occasion. The close relationship between the NY1999 strain and strains isolated from Israel in 1998/2000 led to the conclusion that the source of the 1999 virus was the Middle East (Lanciotti et al. 1999). However, an in-depth analysis of WNV activity in Israel indicated that the Israel1998 WNV was introduced by migrating storks and was not endemic to the region (Malkinson et al. 2002). In that report, it was stated that the migrating storks were infected in Europe, either near the nesting site or along the route of migration, and they introduced the virus into Israel in August of 1998. Our hypothesis postulates that a reservoir of NY1999/Israel1998 existed somewhere in Europe and was the source for both the Israel (and subsequently New York in 1999) and the Argentina WNVs. Additional analysis of WNV strains in Argentina and investigations looking for this reservoir are needed to answer these questions.
Although several genomic differences have been observed among virulent and nonvirulent WNV strains, the glycosylation of an Asn in the envelope protein (E154) is thought to be one of the main determinants for virulence in WNV both in lineages I and II (Beasley et al. 2004, 2005, Botha et al. 2008). A mutation in E156 Ser would result in loss of glycosylation motif. Because Argentine strains do not posses this mutation, the glycosylation site is conserved. This is not surprising because both of these strains were isolated from fatal horse brains. So the presence of virulent WNV strains in a region with relatively high human population density could be determinant for a human outbreak.
Footnotes
Acknowledgments
We thank all of the people who contributed to the identification and follow-up of these cases. We gratefully acknowledge all the INEVH “Dr. J. I. Maiztegui” staff, in particular Pablo Baroni and Verónica Fasciani, for technical assistance. We gratefully acknowledge Amy Lambert for technical assistance.
This study was supported with federal funds from Argentinean Ministry of Health. Dr. Fabbri is in charge of Molecular Virology of the Arbovirus Laboratory at INEVH, Pan American Health Organization Collaborating Centre for Reference and Research on Arbovirus and Hemorrhagic Fever. Her research interest focuses on diagnosis, research, and epidemiology of arbovirus diseases.
Author Disclosure Statement
No competing financial interests exist.