
Abstract
Background:
Bat-borne viruses pose a potential risk to human health and are the focus of increasing scientific interest. To start gaining information about bat-transmitted viruses in Hungary, we tested multiple bat species for several virus groups between 2012 and 2013.
Materials and Methods:
Fecal samples were collected from bats across Hungary. We performed group-specific RT-PCR screening for astro-, calici-, corona-, lyssa-, othoreo-, paramyxo-, and rotaviruses. Positive samples were selected and sequenced for further phylogenetic analyses.
Results:
A total of 447 fecal samples, representing 24 European bat species were tested. Novel strains of astroviruses, coronaviruses, and caliciviruses were detected and analyzed phylogenetically. Out of the 447 tested samples, 40 (9%) bats were positive for at least one virus. Bat-transmitted astroviruses (BtAstV) were detected in eight species with a 6.93% detection rate (95% confidence interval [CI] 4.854, 9.571). Coronaviruses (BtCoV) were detected in seven bat species with a detection rate of 1.79% (95% CI 0.849, 3.348), whereas novel caliciviruses (BtCalV) were detected in three bat species with a detection rate of 0.67% (95% CI 0.189, 1.780). Phylogenetic analyses revealed a great diversity among astrovirus strains, whereas the Hungarian BtCoV strains clustered together with both alpha- and betacoronavirus strains from other European countries. One of the most intriguing findings of our investigation is the discovery of novel BtCalVs in Europe. The Hungarian BtCalV did not cluster with any of the calcivirus genera identified in the family so far.
Conclusions:
We have successfully confirmed BtCoVs in numerous bat species. Furthermore, we have described new bat species harboring BtAstVs in Europe and found new species of CalVs. Further long-term investigations involving more species are needed in the Central European region for a better understanding on the host specificity, seasonality, phylogenetic relationships, and the possible zoonotic potential of these newly described viruses.
Introduction
W
Bats harbor more zoonotic viruses per species than rodents and are now recognized as a significant source of zoonotic agents, some of which are of particular interest because they cause severe human diseases (Luis et al. 2013). Bats often live in large colonies and practice roosting; they fly, travel, and disseminate viruses over considerable distances (Wynne et al. 2013). In the last decades, since the emergence of severe acute respiratory syndrome (SARS) coronavirus and Nipah virus in Asia, Hendra in Australia, and Ebola in Africa, increasing attention has been paid to bats and bat-borne viruses (Chu et al. 2008). Recently, new viruses have been described in different bat species, i.e., rotaviruses (RotV), paramyxoviruses (ParmV), orthoreoviruses (OrthV), and astroviruses (AstV) (Kohl et al. 2012, Kurth et al. 2012, Dacheux et al. 2014, Kemenesi et al. 2014). Information on the ecology and evolution of bat viruses is still scarce, and more extensive surveillance of different bat species from different geographic areas is needed.
In this study, we investigated the occurrence and genetic diversity of bat RNA viruses in Hungary. Bat fecal samples were collected from different geographic areas of Hungary and screened for RNA viruses of six distinct virus families with different sets of consensus primer pairs.
Materials and Methods
Study area, sample collection
Sample collection was performed in several regions of Hungary from a total of 45 sampling locations. All captured bats were identified for species by an experienced chiropterologist according to Dietz and von Helversen (2004). Animals were apparently healthy; there were no visible physiological or clinical manifestations (i.e., unusual behavior, lack of active movement, lethargy). Samples were taken from bats that were captured primarily for bat-banding activities in Hungary. Bats were trapped in 2012 and 2013 by mist nets or harp traps at swarming sites and in their natural foraging habitats. The animals were freed from nets immediately and put into sterile, disposable, highly perforated paper bags individually and were left hanging for a maximum of 30 min to let them defecate; fecal samples were collected from the bags. After sample collection, bats were released at the netting site.
Duplicate sampling was prevented by marking captured bats with paint. All samples were collected in 500 μL of phosphate-buffered saline and kept on dry ice until processed at the laboratory. All bat species in Europe are strictly protected under the Flora, Fauna, Habitat Guidelines of the European Union (92/43/EEC) and the Agreement on the Conservation of Populations of European Bats (
Processing and analysis of samples
After homogenization, samples were centrifuged at 12,000 rpm for 10 min. RNA was extracted from 200 μL of supernatant using a DiaExtract Viral NA Isolation Kit (Diagon) following the manufacturer's instructions. Samples were tested for AstV, coronavirus (CoV), lyssavirus (LyssV), OrthV, RotV, ParmV, and calicivirus (CalV). PCR conditions, primers, and the length of amplicons are shown in Table 1. PCRs were carried out using QIAGEN One-Step RT-PCR Kit (Qiagen) and DiaTaq PCR Kit (Diagon). Positive controls in each reaction were included for all tested viruses, and nuclease-free water was used as negative control. RT-PCR products were analyzed by gel electrophoresis in 2% agarose gel in Tris-borate-EDTA (TBE) buffer stained with GelGreen™. All laboratory procedures with potentially infectious materials were conducted in the BSL-3 laboratory of the University of Pécs, Hungary.
Equimolar amount from each primer.
Cloning, sequencing, and phylogenetic analyses
One-Step RT-PCR amplicons were cloned into a pGEM®-T Easy vector (Promega), and Escherichia coli JM109–competent cells were transformed with the recombinant plasmid. Briefly, E. coli was incubated in Luria-Bertani medium (LB; Sigma Ltd.) supplemented with 100 μg/mL ampicillin as a selective agent. After incubation at 37°C for 20 h, positive clones were selected and the plasmids were extracted using a QIAprep Miniprep Kit (Qiagen). Target amplicons from the positive plasmids were amplified by standard PCR using pGEM®-T Easy Vector–specific primers following the manufacturer's instructions. Amplified DNA products were purified by the QIAquick Gel Extraction Kit (Qiagen) and prepared for sequencing using a BigDye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems). Samples were sequenced bidirectionally on an ABI Prism 310 DNA Sequencer (Applied Biosystems).
Nucleic acid sequences of the new AstV, CoV, and CalV strains were identified preliminarily by GenBank BLAST searches. Further characterization was carried out by phylogenetic analyses with cognate sequences available in public databases. Basic sequence manipulation and verification were performed using GeneDoc v2.7 software. Nucleotide sequences were aligned by ClustalX v2.0 software, and a phylogenetic tree was constructed from the nucleic acid sequence alignments using the maximum likelihood method based on the General Time Reversible model (GTR+G+I) of the program MEGA v5.0 software. The number of bootstrap replications was 1000.
Statistical analyses
The detection rate of different viral infections was estimated using one-way analyses of variance in the prevalence package (v. 0.2.0, Devleesschauwer et al. 2013) of R 3.1.0 software (R Development Core Team 2014).
Results
A total of 447 bat fecal samples were collected in 2012 and 2013 from 45 sampling sites across Hungary (Fig. 1). Twenty-four out of the 28 known Hungarian bat species were sampled and tested in this study, although the number of specimens from different bat species were variable (range, 1–125). All examined bats looked healthy with no detectable disease symptoms. Of the 447 tested samples, 40 (9%) bats were positive for at least one virus and co-infection was observed in a single case (Table 2). Novel strains of AstVs, CoVs, and novel CalVs were detected (Table 2), whereas LyssV, OrthV, ParmV, and RotV were not identified in the samples. Nucleic acid sequences of the new AstV, CoV, and CalV strains were identified preliminarily by GenBank BLAST searches. Further characterization was carried out by phylogenetic analyses with cognate sequences available in public databases.
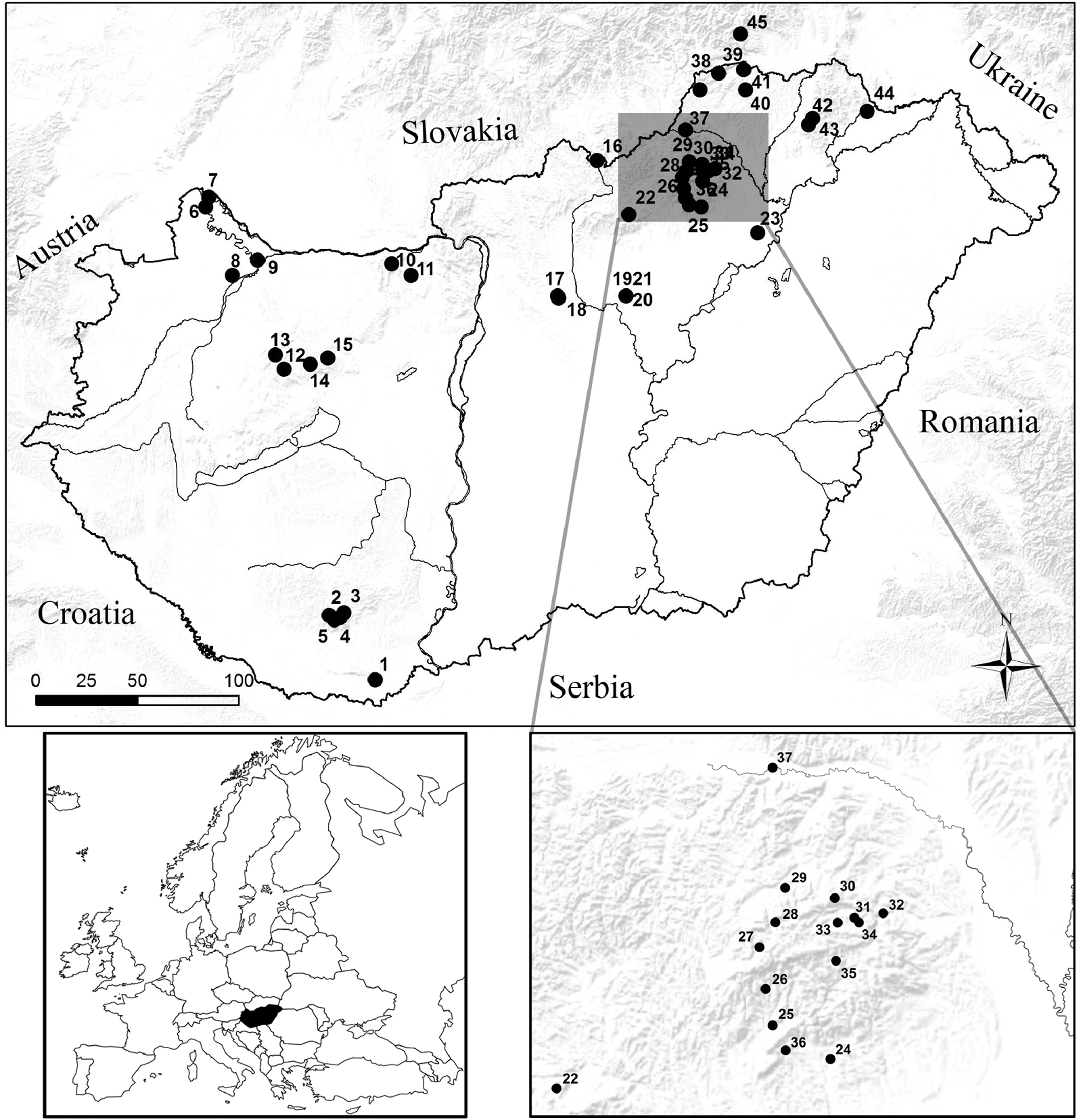
Schematic map of Hungary. Each black dot (•) represents a single sampling site.
The overall detection rate of AstV in bats was 6.93% (95% confidence interval [CI] 4.854, 9.571) with detection rates between 2.7% and 80% per species. Out of the 24 bat species tested, AstVs were identified in the following eight species: Miniopterus schreibersii, Myotis bechsteinii, Myotis daubentonii, Myotis emarginatus, Myotis nattereri, Nyctalus noctula, Pipistrellus pygmaeus, and Plecotus auritus. The detection rates varied significantly between bat species, with M. schreibersii showing significantly higher rates (80%) than any other species (analysis of variance, p=0.001). AstVs were detected in 16 out of the 45 collection sites.
Upon sequence and phylogenetic analysis of a fragment of the RNA-dependent RNA-polymerase (RdRp) gene, the novel Hungarian bat AstV (BtAstV) strains (GenBank acc. nos. KJ652321–KJ652328) clustered with other BtAstV strains identified worldwide, and markedly differed from other mammalian AstVs (Fig. 2). In agreement with previous studies, we also observed a notable genetic variability within the BtAstV strains. Genetically diverse virus sequences were determined from the Myotis spp. (M. daubentonii, M. nattereri, M. emarginatus, and M. bechsteinii), Miniopterus spp., Pipistrellus spp., Plecotus spp., and Nyctalus spp., with patterns of segregation apparently related to the various bat species. Nucleotide identity between the novel BtAstV strains detected in Hungary and other BtAstVs detected worldwide ranged from 51% to 75%. However, the average nucleotide divergence between AstV species recognized by the International Committee on Taxonomy of Viruses (ICTV) was calculated as 55%. On the basis of the low genetic divergence in the RdRp gene, the novel AstV strains identified in the Hungarian bats might represent potentially new species of AstVs.

Phylogenetic analyses of novel astroviruses (BtAstV) identified from bats. The phylogenetic tree was constructed based on a 420-bp-long region of the RNA-dependent RNA polymerase gene. The Hungarian BtAstV strains detected in this study are marked in bold face.
CoV RNA was detected in seven bat species: M. daubentonii, Myotis myotis, M. nattereri, P. pygmaeus, Rhinolophus euryale, Rhinolophus ferrumequinum, and Rhinolophus hipposideros. The overall detection rate of bat CoV (BtCoV) among the sampled bats was 1.79% (95%, CI 0.849, 3.348). Bats were found positive for BtCoVs in seven sampling locations. BtCoV was identified in three European bat genera and seven species. SARS-like CoV (GenBank acc. nos. KJ652335) was detected in the species R. euryale, while alphacoronavirus sequences were obtained from R. ferrumequinum, R. hipposideros, M. daubentonii, M. myotis, M. nattereri, and P. pygmaeus bats (GenBank acc. nos. KJ652329–KJ652334). Co-infection with BAstV and BtCoV was observed in a single case of P. pygmaeus. In the RdRp gene-based phylogenetic analyses (Fig. 3), the novel Hungarian BtCoV strains clustered together with other BtCoV strains from Germany and Bulgaria. The Hungarian BtCoV strains of the alphacoronavirus group displayed 52–96% nucleotide identity to non-Hungarian alphacoronaviruses, whereas the Hungarian betacoronavirus strains displayed 82–96% nucleotide identity to other betacoronaviruses.
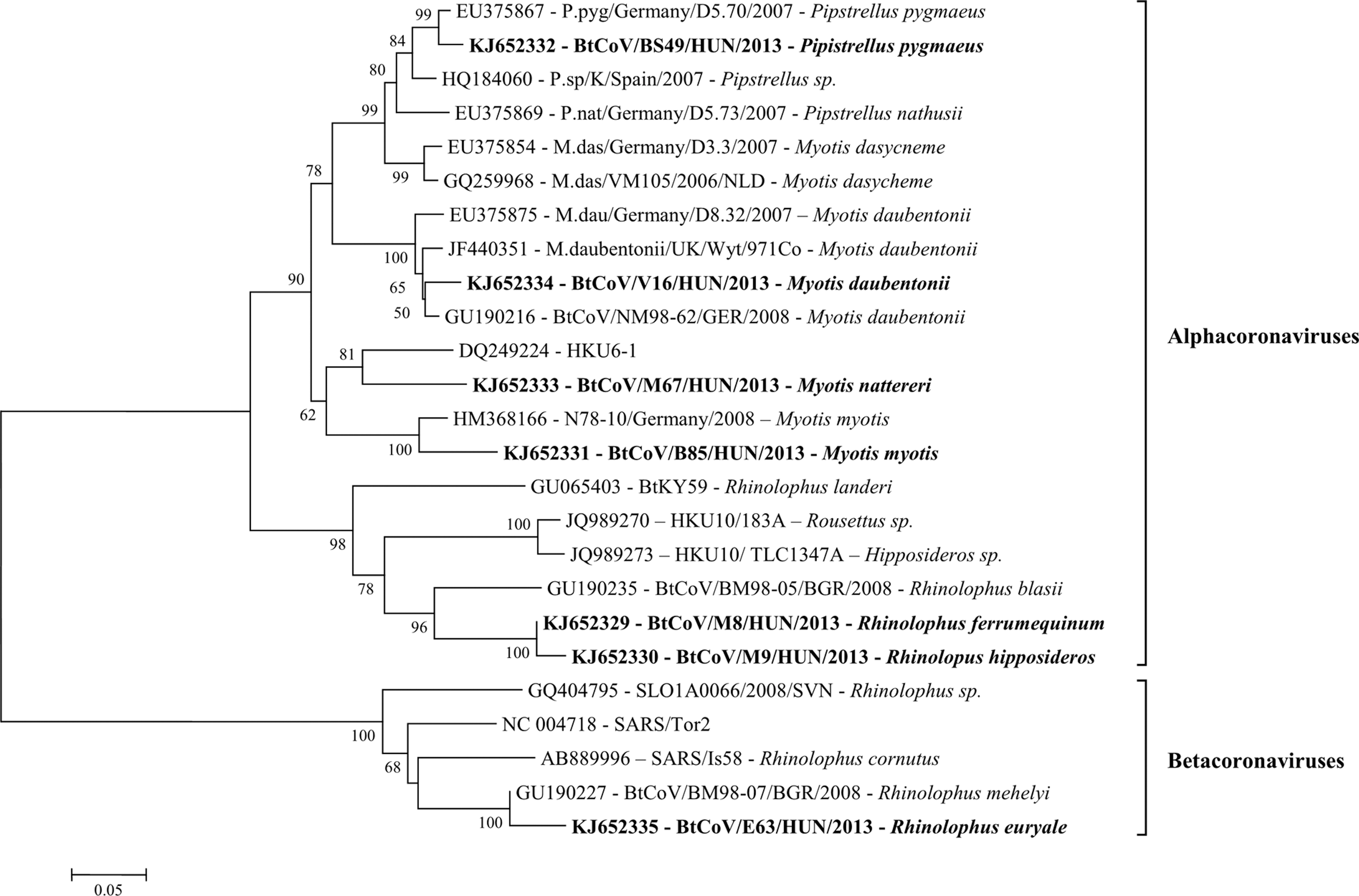
Phylogenetic tree of bat-transmitted alpha- and betacoronaviruses (BtCoV) detected in Hungary. Analyses was performed based on a 440-nucleotide segment of the RNA-dependent RNA polymerase gene. BtCoV strains identified in this study are marked in boldface.
Novel strains of bat CalV (BtCalV) were detected in three bat species, namely M. daubentonii, Myotis alcathoe, and Eptesicus serotinus. BtCalV-positive bats were detected in three locations. The detection rate of BtCalV among the sampled bats was 0.67% (95%, CI 0.189, 1.780). A sequence similarity search using BLASTN against the National Center for Biotechnology Information (NCBI) nonredundant nucleotide database characterized the viruses as members of the Caliciviridae family. Based on the sequence analysis (Fig. 4) of a fragment of the RdRp gene, nucleotide identity between the novel Hungarian BtCalV strains (GenBank acc. nos. KJ652318–KJ652320) and other CalVs ranged from as low as 30% to 56%. However, classification below the family level was not possible, because the Hungarian BtCalV did not cluster with any of the CalV identified in the family so far. Strain BtCalV/M63/HUN/2013 (GenBank acc. no. KJ652319), detected form M. daubentonii, was more closely related to porcine enteric sapoviruses, differing from bat sapoviruses identified in China, suggesting this strain may be a member of the Sapovirus genus. Strain BtCalV/BS58/HUN/2013 (GenBank acc. no. KJ652318), identified from E. serotinus, appeared as an outlier between the genera Recovirus and Valovirus. Even more interesting, strain BtCalV/EP38/HUN/2013 (GenBank acc. no. KJ652320) identified from M. alcathoe, segregated with avian CalV strains, rather than with other mammalian viruses.
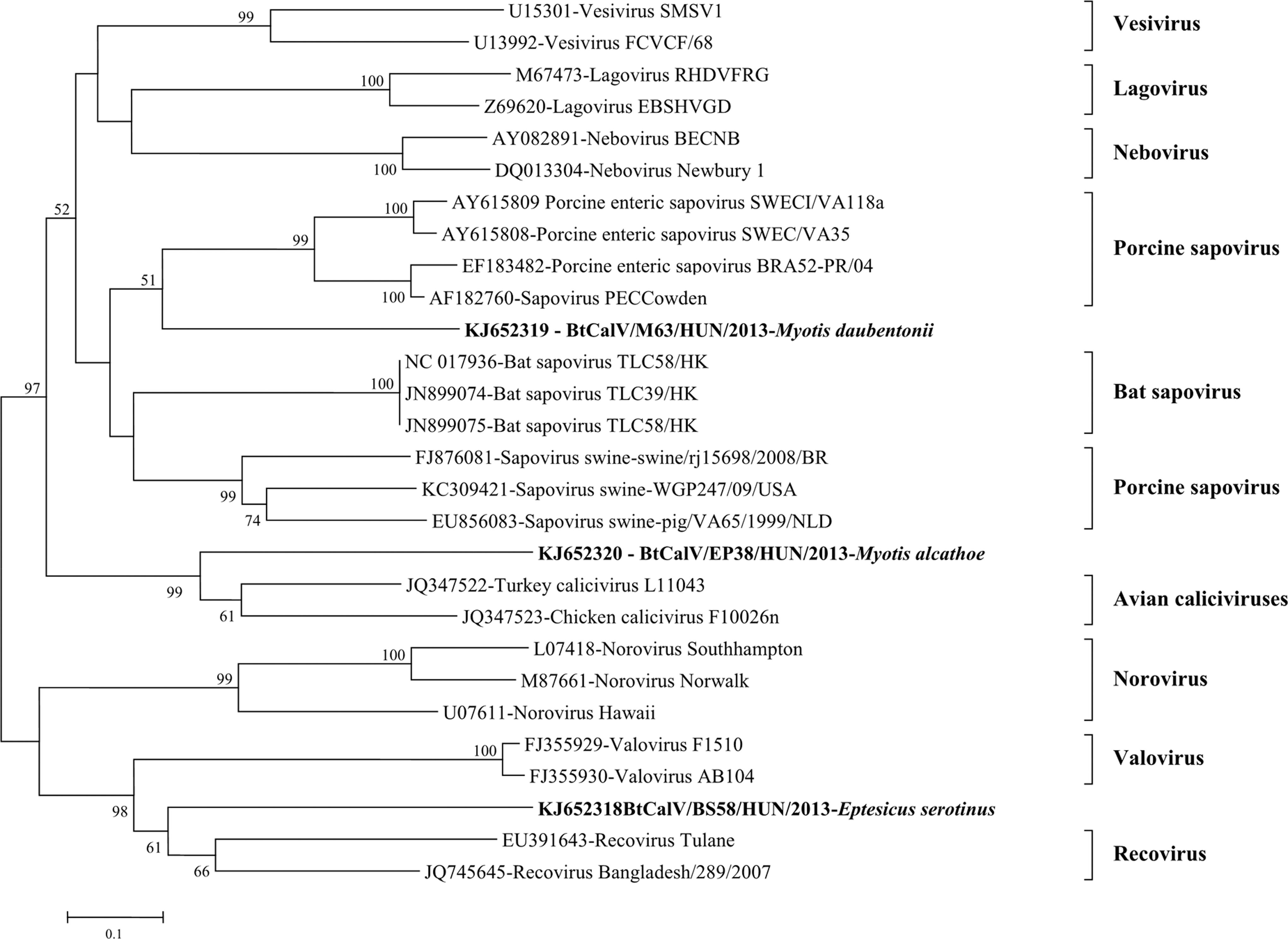
Unrooted maximum likelihood tree of novel bat caliciviruses (BtCalV) om the basis of a partial (320 nucleotide) sequence of the RNA-dependent RNA polymerase gene. Novel BtCalV strains are marked in boldface.
Discussion
The increasing relevance of bat-transmitted viruses in public health is unquestionable as new viruses have emerged in the last decades causing worldwide epidemics. Due to the fact that there are no systematically collected data about the presence of bat-transmitted viruses among Central European bat fauna, we conducted a large-scale surveillance in different geographical locations of Hungary. A total of 447 fecal samples from 24 different bat species were collected and tested for various RNA viruses. Although we were able to detect three out of the six virus families, the sample size limitations of certain bat species might be a possible cause of negative findings. Forty bats were found positive for at least one virus, with one sample containing a mixed infection. Because none of the sampled animals showed evident disease symptoms when captured, our findings indicate that bats can shed several viruses at the same time asymptomatically.
One of the most intriguing findings of our investigation is the discovery of novel BtCalVs in Europe. Thus far, only bat sapoviruses have been published, each from a single Chinese bat species, Hipposideros pomona (Tse et al. 2012). In this study, bat BtCalVs were identified from three different bat species. M. daubentonii and E. serotinus have a broad distribution area across Eurasia. However, the distribution of M. alcathoe is limited to Europe. Strain BtCalV/M63/HUN/2013 segregated with viruses of the Sapovirus genus, although it is genetically unrelated to the Chinese BtCalVs. In contrast, the BtCalV strains BtCalV/BS58/HUN/2013 and BtCalV/EP38/HUN/2013 displayed unique genetic features, as they could not be classified into any of the established CalV genera. Strain BtCalV/BS58/HUN/2013 identified from E. serotinus was a genetic outlier between recoviruses and valoviruses, identified in primates and swine, respectively (Farkas et al. 2008, L'Homme et al. 2009). Even more interesting is that strain BtCalV/EP38/HUN/2013 identified from M. alcathoe appeared to be genetically more related to avian CalV strains than to other mammalian viruses. Full-genome sequencing would help to assess the genetic makeup of these novel CalVs and their possible co-evolution with their putative host species in a greater detail.
Many bat species serve as reservoirs for a variety of CoVs. In recent years, a wide range of CoVs have been detected among European bat species in the United Kingdom, Germany, The Netherlands, Bulgaria, and Slovenia (Gloza-Rausch et al. 2008, Drexler et al. 2010, Reusken et al. 2010, Rihtaric et al. 2010, August et al. 2012). In our study, the overall detection rate of coronaviruses was 1.79%, which is lower than the values reported in other European studies (Table 3). The greater detection rates of CoVs in previous European studies are mainly due to their high detection rates among Myotis dasycneme bats. In Hungary we found no evidence for such a high detection rate in specimens originating from M. dasycneme, although we tested only 11 individuals. Additional investigations involving a greater number of samples collected from M. dasycneme might help resolving this discrepancy.
It has to be noted that the difference in detection rates might be accounted for by the different study design. Due to the large number of bat species we tested, we successfully detected CoVs in seven European bat species within the same geographic area. Most of the sampled animals were captured in natural habitats, but M. myotis, P. pygmaeus, and the three Rhinolophus species may also occur in settlements, because part of the population roosts in buildings. This behavior is becoming frequent due to disturbance of underground roosts (caves and mines) as natural roosting locations (Uhrin et al. 2012). The growing urbanization of these species may provide the ground for a greater frequency of interactions between humans and bats.
The three sets of CoV-specific primers used in our study showed great differences in detection success rates. We found that primers published by de Souza-Luna et al. (2007) were the most appropriate for a primary surveillance of bat-CoVs from fecal samples. This might be explained by the high genomic diversity of CoVs and the different specificity of the primer sets even within the highly conserved region targeted by the primers. Because bats may harbor divergent CoVs highly pathogenic to humans and/or domestic animals (such as SARS and Middle Eastern respiratory syndrome [MERS] coronaviruses), the systematic comparison and further improvements of various diagnostic assays that are suitable to detect potential zoonotic CoVs seem crucial from both public health and veterinary perspectives, as described previously by Memish et al. (2013).
The first report on BtAstVs was published in 2008 (Chu et al. 2008). Since then, only a few additional studies revealed bats as reservoirs of AstVs in Europe and Asia (Zhu et al. 2009, Drexler et al. 2011, Anthony et al. 2013, Kemenesi et al. 2014). Only M. myotis, M. daubentonii, M. bechsteinii, and P. auritus have been addressed before as AstV reservoirs in Europe. Herewith, we have described five new bat species as potential reservoirs for AstVs (Table 2). Although the amplified RdRp gene is the most conserved region of AstV genome, on the basis of the short genomic stretch analyzed, we gathered evidence that multiple lineages of BtAstVs may be co-circulating among Hungarian bat species. The overall detection rate of BtAstVs was 6.94% among our samples, whereas in a Chinese study the detection rate varied between 11.8% and 46% (Chu et al. 2008, Xiao et al. 2011).
This discrepancy might be explained with the different bat fauna and study design. Out of the 30 BtAstV positive samples, 12 originated from Schreiber's bat (M. schreibersii) from a single colony. Schreiber's bat is the only bat species in Hungary that roosts almost exclusively in underground shelters (Gombkötő et al. 2007). These colonies are usually large and dense because they can save considerable amount of energy if their bodies are in close contact during the hibernation period. These bats may roost together with R. ferrumequinum, R. euryale, M. myotis, Myotis blythii, and M. emarginatus. M. schreibersii is one of the fastest flying bats in Europe and can travel large distances (>500 km) from one roost to another (Hutterer et al. 2005).
All of these factors may contribute to the higher detection rate of AstVs in this species, but because all samples originate from only a few locations, we cannot rule out that only these populations have such a high infection rate. The hypothesis that large colonies as in case of M. schreibersii favor the spread of different viruses (i.e., AstVs), is supported also by previous studies in which marked variations were observed in the rates of detection, varying between 0.8% and 36.4%, depending on the geographic area examined (Xiao et al. 2011). In the recent taxonomic nomenclature of ICTV, 19 species of mammalian AstVs have been proposed (Mamastrovirus 1–19), with seven Mamastrovirus species (12 and 14–19) being identified in bats. On the basis of the findings of our study, we assume that potentially newly identified AstV species might circulate among European bat populations. To clarify the taxonomic nomenclature of AstVs, further studies analyzing the complete genome of these viruses are needed.
In the present study, LysVs, ParmVs, OrVs, and RotVs were not detected. These viruses were previously identified in bats from different European countries (Kohl et al. 2012, Kurth et al. 2012, Aréchiga Ceballos et al. 2013, Dacheux et al. 2014). It remains unclear whether these findings are accounted for by bias in sampling, limits of the diagnostics, variations in duration of fecal shedding, or seasonal/geographical differences. It is possible that more complex investigations would be needed to identify these viruses.
Conclusions
To obtain a clearer picture about the prevalence of bat-borne viruses, we carried out a large-scale surveillance of European bat species in Hungary. Fecal samples were tested for multiple virus groups. The main results of this study are successful confirmation of potentially new species of BtCalVs in numerous bat species. Also, we have described new bat species harboring BtAstVs in Europe and found new strains of BtCoVs. Although there are different studies conducted in Europe (Kohl and Kurth 2014), we assume that further long-term investigations involving more species are needed for a better understanding on the host specificity, seasonality, phylogenetic relationships, and the possible zoonotic potential of these new viruses.
Footnotes
Acknowledgments
The research of Ferenc Jakab was supported by the TÁMOP 4.2.4. A/2-11-1-2012 0001–National Excellence Program Elaborating and Operating an Inland Student and Researcher Personal Support System. The project was subsidized by the European Union and co-financed by the European Social Fund. This study was supported by the Hungarian Scientific Research Fund (OTKA; PD77977) project. Krisztián Bányai was supported by the “Momentum program.” Bat netting and ringing activities were partially conducted under the National Biodiversity Monitoring System. We thank Balázs Máté for helping in the field sampling.
Author Disclosure Statement
No competing financial interests exist.