
Abstract
Bartonella infections were investigated in wild rodents from northwestern Chihuahua, Mexico. A total of 489 rodents belonging to 14 species were surveyed in four areas. Bartonella bacteria were cultured from 50.1% of rodent samples (245/489). Infection rates ranged from 0% to 83.3% per rodent species, with no significant difference between sites except for Cynomys ludovicianus. Phylogenetic analyses of the citrate synthase gene (gltA) of the Bartonella isolates revealed 23 genetic variants (15 novel and 8 previously described), clustering into five phylogroups. Three phylogroups were associated with Bartonella vinsonii subsp. vinsonii, B. vinsonii subsp. arupensis, and B. washoensis, respectively. The other two phylogroups were not genetically related to any known Bartonella species. The genetic variants and phylogenetic groups exhibited a high degree of host specificity, mainly at the genus and family levels. This is the first study that describes the genetic diversity of Bartonella strains in wild rodents from Mexico. Considering that some variants found in this study are associated with Bartonella species that have been reported as zoonotic, more investigations are needed to further understand the ecology of Bartonella species in Mexican wildlife and their implications for human health.
Introduction
B
In recent years, several studies have described epidemiological, ecological, and phylogenetic aspects of rodent-associated Bartonella around the world (Bai et al. 2009b, Gundi et al. 2009, Tsai et al. 2010, Jiyipong et al. 2012, Paziewska et al. 2012). In North America, most studies have been in the United States and Canada (Jardine et al. 2005, Jardine et al. 2006, Bai et al. 2007, Morway et al. 2008, Gundi et al. 2012), whereas little information is available from Mexico. To our knowledge, the only investigations on bartonellae associated with rodents or their fleas in Mexico have been conducted in the states of Chihuahua (Bai et al. 2009a) and Sonora (Zapata et al., unpublished). The former study examined the prevalence of Bartonella spp., whereas the latter examined the molecular analysis of Bartonella infection in fleas from rodents. These studies were carried out in northwestern Mexico, but given the small sample sizes of these studies, as well as the lack of information about the genetic diversity of bartonellae circulating in rodents from that region, more investigation is needed. Specific studies in northwestern Mexico become relevant considering that this geographic region has biotic and abiotic features (e.g., rodent community composition, and environmental conditions) that are similar to those of southwestern United States regions, where human cases of rodent-associated Bartonella illness have been reported (Kosoy et al. 2003, Iralu et al. 2006).
The objectives of this study were to: (1) Estimate and compare the prevalence of Bartonella infection (among species, sites, and seasons) in wild rodents from northwestern Mexico by culturing blood; (2) evaluate the genetic diversity of Bartonella strains by analyzing partial sequences of the citrate synthase gene (gltA); (3) analyze host specificity of Bartonella strains; and (4) compare the isolates found in northwestern Mexico with strains of Bartonella that have been reported elsewhere.
Materials and Methods
Trapping localities and sampling
Rodents were live trapped within four areas (≥15 km apart): El Cuervo (30°44′05.65″N, 108°20′06.89″W), Monteverde (30°57′01.88″N, 108°45′25.38″W), Pancho Villa (30°54′06.61″N, 108°27′21.18″W), and Rancho Ojitos (30°46′36.70″N, 108°32′07.43″W). All areas are located in the Janos Region (Janos Biosphere Reserve and neighbor municipalities) in northwestern Chihuahua, Mexico. At each area, six to ten sampling plots were established with at least 600 meters separation from each other. The main trapping periods were in October of 2012 (fall) and late March of 2013 (early spring). Additionally, we included data of some rodents sampled in May of 2012.
Trapping locations encompassed a variety of habitats, including mainly native desert grasslands with and without black-tailed prairie dogs (Cynomys ludovicianus) colonies and shrublands (Prosopis spp.). Small rodents were trapped with Sherman traps (7.6 cm×8.9 cm×22.9 cm; H.B. Sherman Traps, Inc., Tallahasse, FL) baited with a mix of oats and vanilla extract. Black-tailed prairie dogs were live trapped with Tomahawk traps (7.6 cm×8.9 cm×22.9 cm; Tomahawk Live Trap Co., Tomahawk, WI) baited with a mixture of corn, oats, wheat, and molasses. Captured individuals were identified at the species level using taxonomic keys (Anderson 1972, Reid 2006). From each individual, we recorded sex and weight and obtained blood samples from the retro-orbital plexus (small rodents) or femoral vein (black-tailed prairie dogs). Blood samples were taken only once in case an individual was captured multiple times in the same trapping period. After handling, animals were ear tagged (model 1005-3; National Band and Tag Co., Newport, KY) and released at their site of capture. In some instances, individuals were euthanized after sample collection using an overdose of isoflurane (Sofloran, PISA, Hidalgo, Mexico) to confirm species identification and for further tissues analysis. In the field, blood samples were immediately stored in liquid nitrogen and then transferred to a −70°C freezer for storage. For laboratory analyses, samples were shipped on dry ice to the Bartonella Laboratory, Centers for Disease Control and Prevention (Fort Collins, CO).
All procedures for trapping and handling rodents followed the guidelines of the American Society of Mammalogists for the use of wild mammals in research (Sikes et al. 2011) and were approved by the Animal Care Committee of the Veterinary School (Universidad Nacional Autónoma de México) and by the Secretariat of Environment and Natural Resources from Mexico (Permit FAUT-0250).
Bacterial culturing
Whole blood or blood diluted in brain heart infusion medium containing 5% fungizone (Amphotericin B) was plated on agar supplemented with 5% rabbit blood. Each agar plate was divided into four parts, which were inoculated with 10 μL of blood each. Agar plates were incubated aerobically at 35°C in 5% CO2 for up to 4 weeks. Plates were monitored for growth at least once per week after initial plating. Bacterial colonies were tentatively identified as Bartonella species on the basis of colony morphology. Selected colonies were harvested from initial plates or from subsequent subcultures and stored in 10% glycerol for further analysis.
PCR and sequence analyses
Cultures were verified as bartonellae by PCR amplification of a specific region of gltA, using primers BhCS781.p and BhCS1137.n, resulting in a 379-bp product (Norman et al. 1995). Crude DNA extracts were obtained by heating a heavy suspension of the organisms at 95°C for 15 min. The PCR amplifications were performed in a 25-μL reaction mixture containing 200 μM of each DNA template, 12.5 μL of GoTaq® Green Master Mix (Promega, Madison, WI), nuclease-free water, 1 μL each of 10 μM forward and reverse primers, and 2.5 μL of DNA. The PCR was carried out in a TaKaRa Thermal Cycler Dice TP600 (TaKaRa Bio. Tokyo, Japan) or PTC 200 Peltier Thermal Cycler (MJ Research, Watertown, MA), using the following parameters: A 3-min denaturation at 95°C, followed by 35 cycles of 30 s denaturation at 95°C, 30 s annealing at 51°C, and 30 s elongation at 72°C. Amplification was completed by holding the reaction mixture at 72°C for 7 min. The PCR products were visualized for amplicons of the correct size by electrophoresis in a 1.5% agarose gel with ethidium bromide staining. Amplicons of the proper size were purified using a QIAquick PCR Purification Kit (Qiagen, Germantown, MD) and sequenced in both directions using an Applied Biosystems Model 3130 Genetic Analyzer (Applied Biosystems, Foster City, CA). Sequencing reactions were carried out in a PTC 200 Peltier Thermal Cycler (MJ Research, Watertown, MA) using the same primers for PCR assay at a concentration of 3.3 μM. Cycle parameters for the sequencing reactions were 96°C for 1 min, 25 cycles of 96°C for 10 s, 50°C for 5 s, and 60°C for 4 min.
Analysis of sequences and phylogenetic relationships were conducted using MEGA5 (Tamura et al. 2011). The sequences of this study and the known Bartonella species retrieved from the GenBank were aligned using ClustalW. Phylogenetic trees were drawn using the neighbor-joining method by the Kimura two-parameter distance method. Bootstrap calculation was carried out with 1000 replicates. Phylogroups were defined based on the species cutoff proposed by La Scola et al. (2003).
Statistical analysis
We used generalized linear models (GLM) with binomial distribution and logit function to identify the variables that may explain Bartonella prevalence in rodents (the proportion of infected rodents). The explanatory variables analyzed were host identity (rodent species), sites, and trapping period. First, we built a model that included all rodent species that had a sample size greater than 10 and then we built models for each rodent species. For these analyses, we used data from the two main trapping periods (October, 2012, and March, 2013). For individual variables, p values ≤0.05 were considered statistically significant. The overall performance of each model was evaluated with Pseudo-R2 (proportion of explained deviance). The analyses were performed in R software (R Development Core Team, 2014).
Results
Animals
A total of 489 rodent individuals were analyzed to detect the presence of Bartonella infection. Rodents tested represented 14 species belonging to nine genera of three families (Table 1). The most abundant species was the Merriam's kangaroo rat (Dipodomys merriami, 38.7%), followed by the banner-tailed kangaroo rat (D. spectabilis, 16.4%), and the Mearns's grasshopper mouse (Onychomys arenicola, 11.5%) (Table 1).
Bartonella prevalence
Bartonella cultures were obtained from blood samples of 50.1% (245/489) of tested rodents, which belong to 12 rodent species (Table 1). The prevalence of Bartonella infection varied between rodent host species (Table 1). According to the GLM analysis, the prevalence of Bartonella infection was significantly higher in some species (D. merriami, D. spectabilis, O. arenicola, O. leucogaster, Peromyscus leucopus, and P. maniculatus) than in others (C. ludovicianus and C. penicillatus) (Table 2). Also, the overall prevalence was significantly higher in the spring of 2013 (58%) than in the fall of 2012 (43.3%) (Table 2). Although the rodent communities of the four sites varied in species composition, the overall prevalence of Bartonella infection did not differ among them. Intraspecific comparisons showed that infection prevalence did not differ between areas, except for the black-tailed prairie dog, which exhibited a marginally significant lower prevalence in Monteverde (4.2%) compared to the other two areas (∼30%) (Tables 1 and 2). Among the most abundant species (C. ludovicianus, C. penicillatus, D. merriami, D. spectabilis, O. arenicola, and P. maniculatus), only O. arenicola and P. maniculatus species had a significantly higher prevalence of Bartonella in the spring of 2013 than in the fall of 2012 (Table 2).
Results are presented for all species combined and for those species having at least 10 sampled individuals.
p ≤ 0.05.
†Marginally significant.
Genetic diversity
Sequencing of the gltA fragment was performed randomly among 98 isolates obtained from 96 rodents. We identified 23 different genetic variants (Table 3) with 0.3–13.2% divergence among them. Of these variants, 15 were newly identified in the present study with GenBank accession numbers KJ719284–KJ719298. The other eight variants were identical to some previously described genotypes (Table 3). One grasshopper mouse (O. leucogaster) and one kangaroo rat (D. spectabilis) were concurrently infected with two genetically distinct Bartonella variants. The variants from the grasshopper mouse had 10.6% of divergence between them, and the variants from the kangaroo rat diverged in 0.3%.
CH, Chaetodipus hispidus; CP, Chaetodipus penicillatus; CL, Cynomys ludovicianus; DM, Dipodomys merriami; DO, Dipodomys ordii; DS, Dipodomys spectabilis; NA, Neotoma albigula; OA, Onychomys arenicola; OL,Onychomys leucogaster; PL, Peromyscus leucopus; PM, Peromyscus maniculatus; SS, Spermophilus spilosoma.
E, El Cuervo; M, Monteverde; P, Pancho Villa; R, Rancho Ojitos.
Variant previously described elsewhere.
On the basis of the sequence similarities, the 23 variants were clustered into five phylogroups (groups I–V) (Fig. 1). The genetic distances between groups ranged from 3.6% to 13.2%. Group I included five variants with 14 sequences obtained from O. arenicola (10), O. leucogaster (2), and Neotoma albigula (2), which clustered with B. vinsonii subsp. vinsonii (96.5–97.8% similarity). In this group, one variant identified in eight O. arenicola and two O. leucogaster was identical to a variant previously found in O. leucogaster from Kansas (GenBank acc. no. DQ357613; Bai et al. 2007). Group II included three variants from 10 sequences obtained from P. maniculatus (6), P. leucopus (3), and N. albigula (1) that were clustered with B. vinsonii subsp. arupensis (96.5–99.1% similarity). These variants are identical to previously described Bartonella isolates obtained from P. maniculatus in North Carolina and New Mexico and from N. albigula in New Mexico (GenBank acc. nos. AF489539, U84380, U43882, and AF148487; Kosoy et al. 1997).
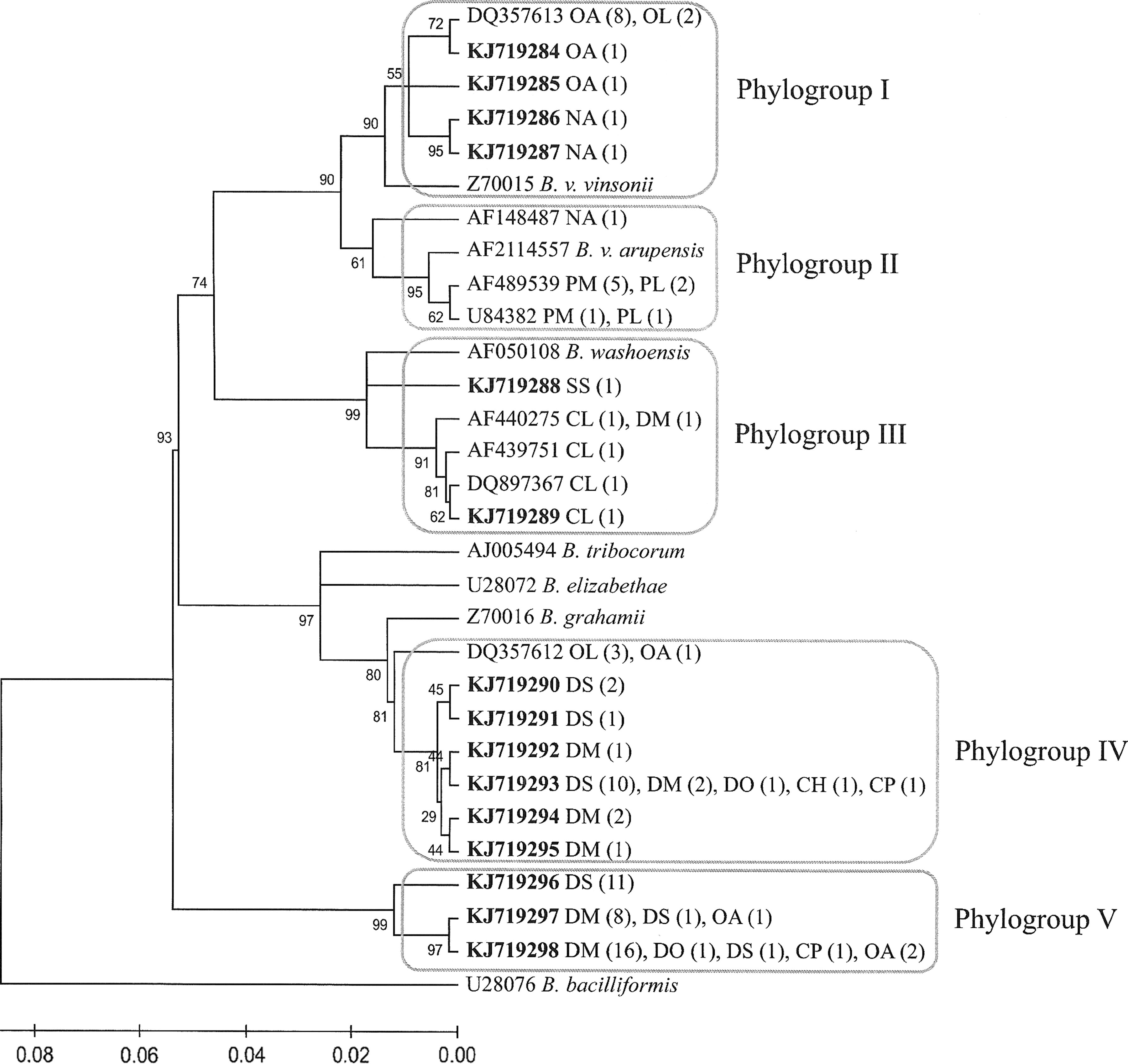
Phylogenetic classification of 98 Bartonella genotypes based on gltA gene sequences detected in 96 rodents from northwestern Mexico. The phylogenetic tree was constructed by the neighboring-joining method, and bootstrap values were calculated with 1000 replicates. Each genotype is indicated by its GenBank accession number, with novel genetic variants in boldface. After each accession number is the rodent species from which Bartonella isolates of the genotype were obtained. Numbers in parentheses are the number of isolates obtained from rodents of the indicated species. Species abbreviations are in Table 3. The sequences were classified into five phylogroups, each indicated by a rectangle. A criterion of ≥96% homology to gltA was used to define phylogroups (La Scola et al. 2003).
Group III clustered with B. washoensis (96.5–96.8% similarity) and included five variants from six sequences, mainly from ground squirrels (four C. ludovicianus and one Spermophilus spilosoma), but also from one kangaroo rat (D. merriami). Three of these variants were identical to variants obtained from Bartonella isolates of C. ludovicianus (GenBank acc. nos. AF440276, AF440276, DQ897367; Kosoy et al. 2003, Stevenson et al. 2003, Bai et al. 2008) and their fleas (GenBank acc. no. AF440732; Stevenson et al. 2003) in Colorado. Group IV included seven variants. One variant was represented by sequences obtained from one O. arenicola and three O. leucogaster. Four variants were obtained from either D. merriami or D. spectabilis, whereas one variant was found in sequences of five heteromyid species: D. spectabilis (10), D. merriami (2), D. ordii (1), Chaetodipus hispidus (1), and Chaetodipus penicillatus (1). Another variant was identical to the sequence described previously in O. leucogaster from Kansas (GenBank acc.no. DQ357612; Bai et al. 2007). The closest Bartonella species to this group is B. grahamii with 97.1–97.8% similarity. Group V consisted of three variants—one variant from 11 sequences of D. spectabilis; one variant from eight D. merriami, one D. spectabilis, and one O. arenicola; and one variant from four heteromyid species (16 D. merriami, one D. ordii, one D. spectabilis, and one C. penicillatus), and two O. arenicola. No variant from this group has been previously described.
Discussion
The overall prevalence of Bartonella in rodents from the region of Janos (∼50%) was comparable with reports from other regions of North America (Kosoy et al. 1997, Jardine et al. 2005, Bai et al. 2007) and varied significantly among host species, which is similar to the pattern observed in other areas. For example, the high prevalence reported in this study for Onychomys and Peromyscus (∼40– 80%) falls within the range observed in the United States (Kosoy et al. 1997, Bai et al. 2007, Bai et al. 2011). Similarly, the low prevalence found in Chaetodipus spp. (∼12%) is in agreement with the prevalence of Bartonella reported for these rodents in the United States (Bai et al. 2007, Bai et al. 2009a). On the other hand, the lack of spatial variation among sites in the prevalence of Bartonella found in this study is similar to the pattern observed in other studies from North America (Holmberg et al. 2003, Jardine et al. 2005). The only difference in prevalence between sites was observed in C. ludovicianus. However, further studies are required to confirm this observation, considering that the sample size was relatively small (13–24 individuals per study area) and the result was marginally significant. Previous studies in more northern locations have reported a higher prevalence of Bartonella in rodents during the late summer and fall than other seasons (Fichet-Calvet et al. 2000, Kosoy et al. 2004, Jardine et al. 2006). Different from that, the overall prevalence in this study was higher in early spring than in fall. More extensive long-term studies are necessary to have a better understanding of the temporal dynamics of Bartonella in this region.
Phylogenetic analyses of the gltA sequence obtained from Bartonella cultures revealed that rodent communities from the Janos region harbor diverse Bartonella strains representing a variety of species. The five phylogroups detected indicated at least five species or subspecies of Bartonella. Groups I, II, and III are associated with previously described Bartonella species or subspecies from North America: B. vinsonii subsp. vinsonii, B. vinsonii subsp. arupensis, and B. washoensis, respectively (Kosoy et al. 1997, Jardine et al. 2005, Bai et al. 2007, Bai et al. 2008, Bai et al. 2011). Two of these species (B. vinsonii subsp. arupensis and B. washoensis) have been described as zoonotic pathogens (Welch et al. 1999, Kosoy et al. 2003). This warrants further study to understand their potential role in causing human diseases in Mexico. The remaining groups (IV and V) may represent new species of Bartonella; however, further studies are required to describe this group fully.
Although a portion of the gltA gene is commonly used for identifying Bartonella isolates from rodents (Buffet et al. 2013), we realize that this is not a sufficient approach for characterization of Bartonella species. It was used in our study only for comparing the obtained isolates with other rodent-associated Bartonella species. A genomic analysis of the obtained strains would be desirable in the future.
Host specificity of Bartonella species has been described in North American rodents from field and laboratory studies (Kosoy et al. 1997, Kosoy et al. 2000, Jardine et al. 2005, Bai et al. 2007, Bai et al. 2011), whereas in some other parts of the world, specifically in Europe, this is not a clear pattern (Buffet et al. 2013). Our phylogenetic analysis of Bartonella sequences clearly demonstrated an evident pattern of host specificity, particularly at the rodent genus level (genetic variants) and rodent genus or family level (phylogenetic groups). Group I was composed by variants found in grasshopper mice (Onychomys spp.) and N. albigula. Group II is associated with B. vinsonii subsps. arupensis, which is specific to mice of the genus Peromyscus (Bai et al. 2011). Group III included variants clustered with B. washoensis, which has been shown specific to sciurid rodents (Kosoy et al. 2003, Stevenson et al. 2003, Bai et al. 2008, Inoue et al. 2011). Variants from heteromyid rodents (Dipodomys spp. and Chaetodipus spp.) were clustered in groups IV and V.
Although some variants from heteromyid rodents were shared between more than one species, there is a certain degree of host specificity for variants from the most common heteromyid species (D. merriami and D. spectabilis). Some variants associated with heteromyid rodents were also found in O. arenicola. A similar pattern was observed by Bai et al. (2007), who suggested that O. leucogaster is an occasional host of some Bartonella strains that are specific to other rodent species. In that study, the authors hypothesized that this observation can be explained by specific behavior of grasshopper mice that use borrows from other rodent species and also prey on them occasionally. In this study, the infection of a kangaroo rat (D. merriami) with a variant specific to C. ludovicianus might represent another example of spillover of a Bartonella strain between rodent species.
Multiple genotypes of Bartonella have been found in a single animal host (Birtles et al. 2001, Jardine et al. 2005). In the present study, two samples, one from O. leucogaster and one from D. spectabilis, tested positive to two different variants, providing evidence that simultaneous infection with multiple strains of Bartonella can occur in individuals within the rodent community from northwestern Mexico. Eight out of 23 genotypes detected in this study were identical to Bartonella genotypes found in the same rodent hosts (genus or species) from different regions of the United States, which is in agreement with other studies showing that certain Bartonella strains can be distributed across large geographic areas (Jardine et al. 2005), even in different continents (Ellis et al. 1999), according to the distribution of their hosts (Buffet et al. 2013).
To our knowledge, this is the first study on the genetic diversity of Bartonella strains from Mexican rodents. Because some variants found in this study were associated with Bartonella strains that can cause human illness, and because the number of Bartonella species described as zoonotic pathogens is increasing, it is important to extend investigations on Bartonella strains circulating in Mexican wildlife to understand their dynamics and to identify potential risks for human populations.
Footnotes
Acknowledgments
Funding for the study was provided by CONACyT project no. 179482, the Scott Neotropical Fund Award (Cleveland Metroparks Zoo and the Cleveland Zoological Society), and the CDC Global Diseases Detection program. The authors thank A. Fernández, A. Vigueras, S. González, P. Martínez, M. Verona, A. López, and K. Moreno for assistance during field sampling. We are grateful to J. Diaz, E. Ponce, and R. Sierra (Janos Grassland Biological Station, IE-UNAM) for logistical support in the field. A.V Rubio is supported by a CONICYT Becas-Chile Scholarship. G. Suzán acknowledges the support provided by DGAPA-UNAM and CONACyT.
Author Disclosure Statement
No competing financial interests exist.