
Abstract
Previously, we identified five Leishmania mexicana antigens reacting with antibodies from cutaneous leishmaniasis patients, designated on the basis of their molecular weights as p26 (pI 7.8), p27 (pI 8.1), p28 (pI 8.6), p29 (pI 8.5), and p31 (pI 9.0). Among these antigens, p29 was most strongly recognized by the antibodies. Thereafter, p29 was identified as elongation factor-1α (EF-1α) of Leishmania mexicana through mass spectrometry analysis and western blot using a commercial antibody that reacted with EF-1α from different species. Our results showed that the p29 antigen of Leishmania mexicana is EF-1α.
Introduction
L
Previously, we identified five immunodominant antigens, with molecular weights and isoelectric points of 26 kD (pI 7.8), 27 kD (pI 8.1), 28 kD (pI 8.6), 29 kD (pI 8.5), and 31 kD (pI 9.0), in a crude extract of L. mexicana (designated ECLm) that were recognized by the sera of CL patients in two-dimensional western blotting experiments (Salazar-Mejía et al. 2010). Because p29 was the most strongly recognized antigen, we performed proteomic analysis to identify this molecule. The results showed that the p29 antigen is elongation factor-1α (EF-1α) of L. mexicana.
Materials and Methods
L. mexicana strain MHOM/MX/92/UAY68 was generously donated by Dr. Patricia Talamás (Cinvestav-IPN). Preparation of the crude extract of L. mexicana (ECLm) was carried out following the protocol described by Salazar-Mejía et al. (2010). The parasites were cultured as promastigotes at 25°C in RPMI-1640 culture medium supplemented with 5% (vol/vol) heat-inactivated bovine fetal serum and gentamicin (50 μg/mL). ECLm was prepared from 1×108 cells. Briefly, parasite pellets were suspended in a buffer containing urea (8 M), CHAPS detergent (2% wt/vol), and a protease inhibitor cocktail. The suspension was sonicated and centrifuged, and the supernatant was collected. The proteins were precipitated with acetone (80% vol/vol), and the pellet was dissolved in 2D rehydration buffer. The protein concentration in the ECLm was determined by the Bradford method following standard protocols, and the ECLm was stored at −20°C until use.
Isoelectric focusing (IEF) was performed using immobilized pH gradient (IPG) strips with pH values ranging from 3 to 10. A 100-μg sample of the ECLm was loaded onto the strip and incubated overnight at 20°C, following the manufacturer's instructions. The samples were run for 9 h in a Protean IEF System using the following program: (1) 20 min at 250 V, (2) 2 h at 4000 V, and (3) increasing voltages from 4000 to 10,000 V/h. The strips were stored at −70°C until use. For two-dimensional electrophoresis, the strips were resolved on 12.5% sodium dodecyl sulfate (SDS) polyacrylamide gels, as described previously by Salazar-Mejía et al. (2010).
The proteins were then detected with Coomassie G-250 stain, after which the p29 spot was excised and transferred to a siliconized Eppendorf tube. The protein sequence of the band was determined using liquid chromatography in an Accela system coupled to an LTQ-Orbitrap Velos mass spectrometer (LC-MS) at the IBT (Instituto de Biotecnología, UNAM, Cuernavaca, Morelos, México). A duplicate of the unstained two-dimensional gel containing the antigenic proteins (ECLm) was transferred to a 0.22-μm nitrocellulose membrane for western blot analysis, according to the methods of Salazar-Mejía et al. (2010), with some modifications. The nitrocellulose membranes were first blocked with 5% (wt/vol) nonfat milk and then incubated either with serum from a patient with a parasitological diagnosis of LC or with a commercially available antibody specific to human EF-1α (AV48166, Sigma-Aldrich Mexico); both sera were diluted 1:100. After washing, the membranes were incubated with anti-human immunoglobulin G (IgG) conjugated to horseradish peroxidase (HRP) or anti-rabbit IgG-HRP for the blots produced using patient sera or commercial anti-EF-1μ, respectively. Finally, the reaction was visualized using diaminobenzidine and hydrogen peroxide (H2O2). The one-dimensional western blot was developed using an antigenic extract of L. mexicana prepared in more or less aggressive conditions using TGS buffer consisting of Tris (25 mM), glycine (192 mM), and SDS (0.1% wt/vol), without sonication steps. Extracts of RAW 264.7 murine macrophages and THP1 human monocytes prepared in TGS buffer were also included. Reactivity was demonstrated under conditions similar to those described above.
Results and Discussion
Proteomic analysis of the p29 antigen revealed
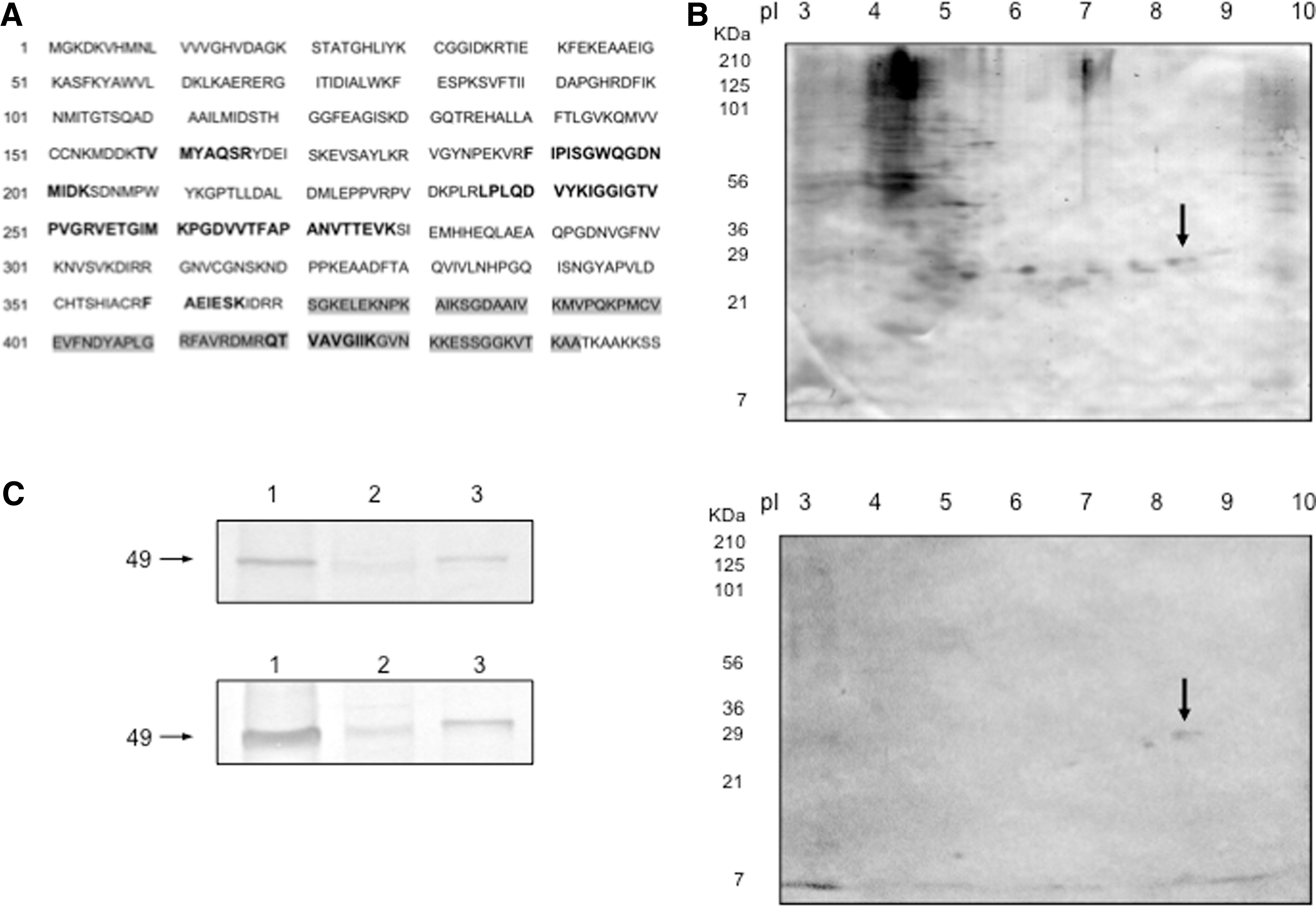
The p29 antigen is elongation factor 1α (EF-1α) of L.mexicana. (
To further confirm the identity of the p29 antigen as EF-1α from L. mexicana, using a two-dimensional western blot of the ECLm, we compared the molecular weight and isoelectric point of the spots detected by sera from a parasitologically confirmed CL patient (Salazar-Mejía et al. 2010) and those detected by a commercial polyclonal antibody against the EF-1α epitope that is conserved among chickens, dogs, pigs, humans, mice, and rats (highlighted with a gray background in Fig. 1A). The patterns of the two-dimensional western blots showed that p29 was recognized by both the patient sera and the commercial antibody against EF-1α (Fig. 1B). Other antigenic spots observed in Figure 1B with a pI lower than 7 showed heterogeneous patterns among patients, as described previously (Salazar-Mejía et al. 2010), and for this reason, these spots were not considered in further analysis. In addition, the lower-molecular-weight protein (p27) detected in the western blot developed with the commercial antibody against EF-1α was sequenced via mass spectrometry and was also identified as EF-1α (data not shown). The low-molecular-weight spots (p29 and p27) observed in the two-dimensional western blot suggested either the presence of different isoforms in Leishmania or the generation of a mechanical product due to sonication of the sample. The latter possibility is most likely because when the western blot was developed using extracts of mammalian cells and L. mexicana obtained by lysis with TGS buffer, both p27 and p29 could not be detected, and instead, an antigen of 49 kD, corresponding to the molecular weight of EF-1α was detected in all cases. Moreover, this antigen was detected using either CL patient sera or the commercial antibody (Fig. 1C). This result may have occurred because the less aggressive conditions applied to prepare the extracts in addition to the reduced contact area in conventional western blotting could have hindered the observation of other epitopes.
Proteomic analyses of Leishmania antigens can provide relevant information regarding the identification of molecules that can be used to improve diagnostic methods or can be employed to determine targets for chemotherapeutic drugs. In a previous study, we identified five immunodominant antigens recognized by sera from confirmed CL patients in a crude extract from L. mexicana promastigotes (ECLm) through two-dimensional western blot analysis. Among these five major antigenic proteins, p29 was selected and analyzed in this study. LC-MS analysis following trypsin digestion of the p29 spot obtained through two-dimensional gel electrophoresis revealed that the amino acid sequence of this protein was 100% identical to that of EF-1α from L. mexicana. EF-1α of Plasmodium falciparum is also known to be antigenic in malaria patients (Costa et al. 2013). However, this antigen may present limited utility in the immunodiagnosis of cutaneous leishmaniasis in Mexico, as suggested by the BLAST analysis of the obtained amino acid sequence that revealed an identity of greater than 92% in alignments with EF-1α proteins from other kinetoplastid parasites that are endemic in Mexico, such as L. braziliensis, L. infantum, and Trypanosoma cruzi, and thus, cross-reactivity may be common.
In normal eukaryotic cells, EF-1α is a guanosine triphosphate (GTP)-binding protein that associates with aminoacyl-tRNA and is involved in the elongation of nascent polypeptide chains at the ribosome. Although the role of EF-1α in cells infected with L. mexicana has not been reported, it may be similar to that of the orthologous protein in Leishmania donovani (Nandan et al. 2002, 2003, 2005, Lopez et al. 2007), which is a species that causes VL in the Old World. The L. donovani homologous EF-1α has been described as an antigenic protein with dual functions; in addition to the canonical function in eukaryotic cells described above, this protein plays an unconventional role in the host–parasite relationship, acting as an activator of Src homology region 2 domain-containing phosphatase-1 (SHP-1 phosphatase) and affecting the macrophage microbicidal activity against Leishmania (Nandan et al. 2002, 2003, 2005). Furthermore, EF-1α from L. donovani is considered to represent a pharmacological target because it contains one structural region that is not exposed in human EF-1α (Lopez et al. 2007). Further studies may lead to the development of drugs that will improve the toxic antimonial therapy currently used against L. mexicana (Nandan et al. 2002, 2003, 2005, Lopez et al. 2007). In conclusion, the results obtained of immunoproteomic analysis of the p29 antigen showed that this protein is the EF-1α of L. mexicana.
Footnotes
Acknowledgments
The authors acknowledge QFB Javier Torrecillas for technical laboratory assistance. This work was supported by the Programa de Fomento y Apoyo a Proyectos de Investigación (PROFAPI/091) of the Universidad Autónoma de Sinaloa. A special acknowledgement goes to Dr. Yukifumi Nawa for critical review.
Author Disclosure Statement
No competing financial interests exist.