
Abstract
The Bunyaviridae family is currently composed of five genera, including Phlebovirus, in which several phleboviruses are associated with human diseases. Using high-throughput sequencing, we obtained and characterized one complete genome of the Arumowot virus (AMTV) isolated in 1978 from Turdus libonyanus, the Kurrichane Thrush, in the Central African Republic (CAR). The genomic segment of the new strain of AMTV isolated in the CAR had 75.4–83.5% sequence similarity and 82–98.4% amino acid similarity to the prototype sequence of AMTV. The different conserved proteins of the small (S) and large (L) segments (Nc, NSP, and RNA polymerase) showed close similarity at the amino acid level, whereas the polyprotein of the medium (M) segment was highly divergent, with 18% and 37.7%, respectively, for the prototype sequence of AMTV and the Odrenisrou virus (ODRV) isolated from Culex (Cx.) albiventris mosquitoes in the Tai forest, Ivory Coast. Phylogenetic analysis confirmed the sequence homology analysis and indicated that AMTV-CAR clustered into the Salehabad virus antigenic complex. The two closest viruses were the prototype sequences of AMTV originally isolated from Cx. antennatus mosquitoes and ODRV. These molecular data suggest the need for a deep genetic characterization of the diversity of this viral species to enhance its detection in the Central African region and to understand better its behavior and life cycle so that its potential spread to the human population can be prevented.
Introduction
T
We describe here a new strain of the Arumowot virus (AMTV). The first strain of AMTV was isolated on the Nile in Sudan in 1963 from Culex (Cx.) antennatus mosquitoes (Berge 1975). In the Central African Republic (CAR), two strains of AMTV were isolated from rodents (Lemniscomys striatus) in 1971 (AnB651d) and in 1978 (AnB2798). In the same year (1978), another strain (AnB7211d) was isolated from a species of bird (Turdus libonyanus, Kurrichane Thrush) (for more information on the virus isolation, see
Recently, Palacios et al. (2013) described the genetic and antigenic relationships of several viruses belonging to the Salehabad species complex of the genus Phlebovirus (Palacios et al. 2013), and since then it was proposed that AMTV belongs to Salehabad virus species complex. Only four other sequences belonging to the Salehabad virus species complex were available at that time and were compared with other sequences related to the genus Phlebovirus in Bunyaviridae. No human cases of diseases due to this virus have yet been reported, although antibodies to AMTV have been found in African populations (Tesh et al. 1976) and in domestic animals, mostly cattle (Akakpo et al. 1989, 1991).
Historically, the Institut Pasteur in Bangui, CAR, has developed past research projects on arboviruses in which several viruses were isolated by cell culture from patients suffering from different syndromes or from mosquitoes and animal reservoirs. Some of these cultures led to cytopathic effect (CPE), and the isolates were roughly identified by immunofluorescence. Recently, several strains of Zika virus (ZIKV) or Middelburg virus (MIDV) have already been characterized using broad-spectrum molecular techniques (Berthet et al. 2014, Tricou et al. 2014). In this study, we describe and characterize genetically a new strain of AMTV (named AnB7211d in the CRORA database
Materials and Methods
Blood samples from the birds were cultured in Vero cells, and serological characterization with mouse immune ascitic fluids (IAF) was performed on pelleted cells. This indicated the presence of an unclassified phlebovirus. For molecular characterization, RNA was extracted from a brain biopsy of a dead mouse with aQIAmp Viral RNA Mini Kit (Qiagen) according to the manufacturer's instructions. Extracted total RNA was treated with Turbo DNase (Life Technologies) to remove the Mus musculus DNA genome and then retrotranscribed into cDNA with Super Script III reverse transcriptase and random hexamer primers (Life Technologies). The cDNA generated was amplified with the Phi29 enzyme, as described previously (Berthet et al. 2008). Amplified DNA was quantified in the Quant-iT Assay (Invitrogen), and a fixed amount of amplified DNA was sequenced with an Illumina® HiSeq 2000 sequencer.
A total of 107 reads with 100 bases were obtained for each sample. All reads were filtered according to quality, and those corresponding to the mouse genome sequence were filtered with Bowtie 2.0 software with the M. musculus Mn10 sequence as reference. The viral reads corresponding to the AMTV genome were selected by a similarity approach with BLASTN and BLASTX search tools and the two complete sequences available in GenBank of AMTV (HM566143 to HM566145) and Odrenisrou virus (ODRV) (HM566173 to HM566175). For each selected read, only the region that matched the viral genome was considered. All reads were initially assembled with ABYSS software with a value of k = 50, and the contigs were then assembled into a “super assembly” with the CAP3 program to obtain the full length of each of three segments of viral genome.
Results and Discussion
Totals of 469,405, 2,648,139, and 1,689,689 reads, representing 3.4%, 19.0%, and 12.1% of all reads, were obtained for the S, M, and L segments, respectively. The average coverage of each genomic segment was 7556×, 5480×, and 8346× for the S, M, and L segments, respectively. Genomic analysis of the AMTV variant isolated in the CAR showed the typical organization of the genome of phleboviruses, with an S segment encoding the N protein and in ambisense orientation a nonstructural protein, an L segment encoding the RNA polymerase, and an M segment encoding a polyprotein (GenBank acc. nos. KJ782449–KJ782451).
As seen in Table 1, the genomic segment of the new strain of AMTV isolated in the CAR had 75.4–83.5% sequence similarity and 82–98.4% amino acid similarity to a previously described sequence of AMTV (strain Ar 1286-64), which originated and was isolated in Spain (Palacios et al. 2011). The different conserved proteins of segments S and L (Nc, NSP, and RNA polymerase) showed close similarity at the amino acid level, whereas the polyprotein of segment M was highly divergent, with 18% and 37.7%, respectively, with AMTV and ODRV originally isolated from Cx. albiventris mosquitoes in the Tai forest, Ivory Coast (Palacios et al. 2011) (Table 1). This high divergence observed for the polyprotein could be due to natural evolution of the strain inside a novel host and by the interaction between external viral proteins and host proteins.
S, small; M, medium; L, large; AMTV, Arumovot virus; ODRV, Odrenisrou virus; SALV, Salehabad virus; ARBV, Arbia virus; ADAV, Adana virus; SRNV, Serra Norte virus; UURV, Urinama virus; ORXV, Oriximina virus; ITAV, Itaituba virus.
A selected set of phlebovirus sequences available from GenBank was used to determine the phylogenetic relations of our novel AMTV variant with other phleboviruses. Sequences were aligned with the ClustalX algorithm in Geneious software, version 6.1.3, at the amino acid level, with manual editing to increase the quality of the alignment. The phylogenetic tree was generated by the neighbor-joining method, and the statistical significance of the tree was estimated by the bootstrapping approach with 1000 replicates. All bootstrapping values are shown as percentages. Phylogenetic analysis indicated that AMTV-CAR (AnB7211d) clustered into the Salehabad virus antigenic complex, and the two closest viruses were AMTV previously described by Palacios et al. and originally isolated from Cx. antennatus mosquitoes and ODRV (Table 1 and Fig. 1). Moreover, no incoherent branching of our AMTV variant was observed for the three segments, suggesting that no reassortment among different species was at the origin of this genome (Fig. 1).
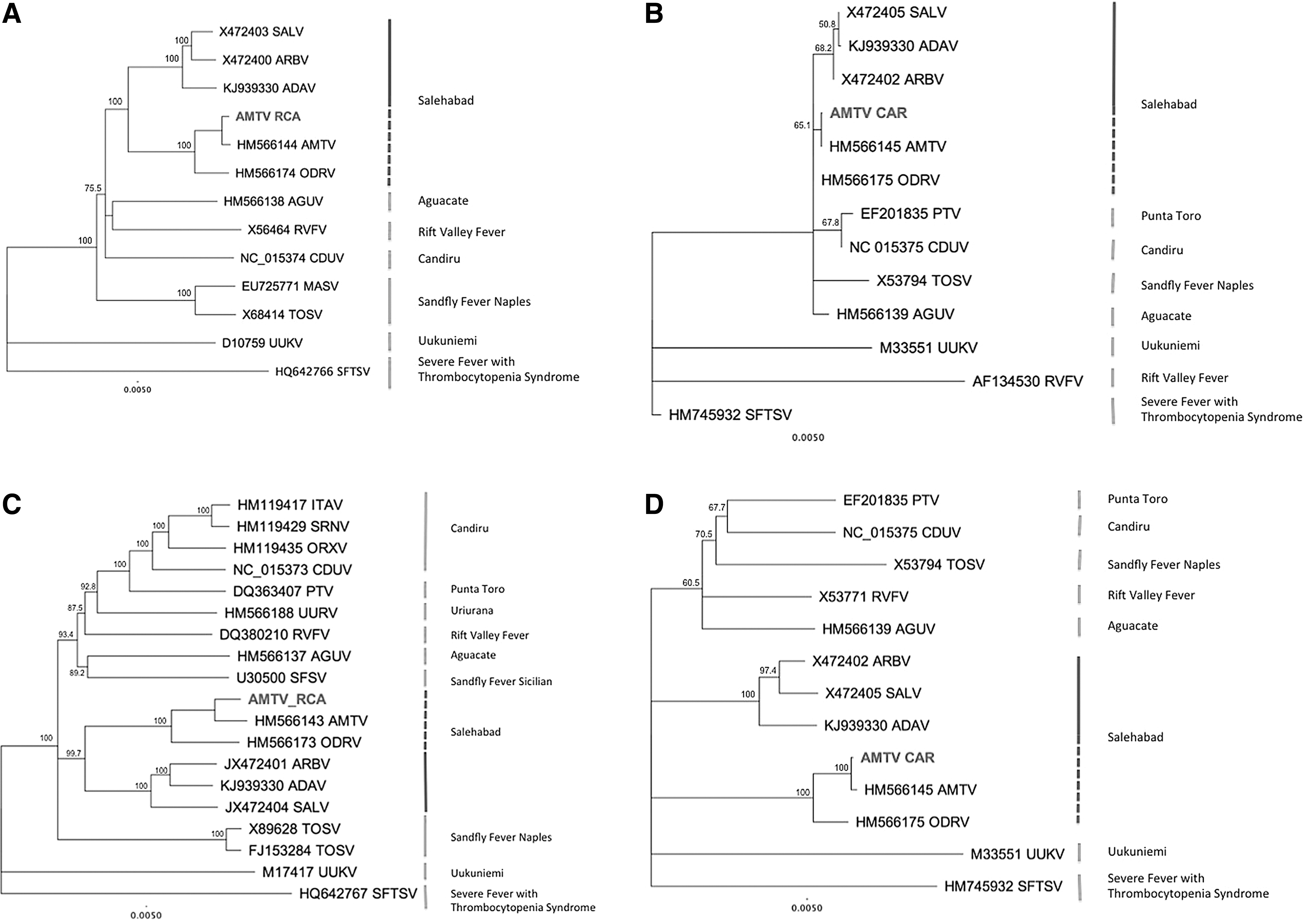
Phylogenetic tree of selected phlebovirus nucleoprotein (
During the past few decades, AMTV has been identified and isolated in various hosts in African countries, such as rodents and mosquitos in Nigeria, Senegal, South Africa, Sudan, and Zimbabwe and cattle, sheep, and goats in Burkina Faso. In spite of its wide circulation, no evidence of pathogenesis was observed in the different hosts. In the CAR, AMTV, like other bunyaviruses (M'poko and Bobaya viruses), was isolated for the first time in T. libonyanus, a species of bird belonging to the Turdidae family. Indeed, the Kurrichane Thrush, whose natural habitat is dry Savanna, is a species of bird largely present in Central African countries, such as the Democratic Republic of Congo, Tanzania, Angola, and other countries in Africa. The CAR has borders in the north with Sudan and Chad. This virus was initially isolated in Sudan from Cx. antennatus mosquitoes (Berge 1975), and no isolation of this virus was done from our large samples of mosquitos collected in Central Africa during several decades. The hypothesis that the bird was infected via an arthropod vector could be not confirmed.
The complete genomic sequence of this AMTV strain suggests that it is genetically close to the strain isolated from Cx. antennatus in 1963 that has been circulating in Sudan (Palacios et al. 2013). In spite of a wide divergence at the nucleic acid level, these strains are close at the amino acid level, except for external viral proteins.
To conclude, this study demonstrates the relevance of high-throughput sequencing both in obtaining the genome and the molecular characterization of a new viral strain. The data generated better established the viral taxonomy, in particular, when the nucleic sequence of the new strain was very distant from the prototype. Moreover, as demonstrated by Alkan et al. (2015), these molecular data suggest the need to improve our knowledge of the diversity of this specific viral species and other members of the Phlebovirus genus, to enhance their detection in the sub-Saharan African region, to understand better their behavior and life cycle, and to prevent their potential spread to the human and animal populations (Alkan et al. 2015).
Footnotes
Acknowledgments
This study was supported by the Institut Pasteur de Bangui and the Institut Pasteur, Paris, France (Programme Transversal de Recherche CEVACAR no. 385). The funders had no role in study design, data analysis, or preparation of the manuscript.
N.B., E.N., A.G., J.C.M., and M.K. conceived and designed the experiments. N.B. performed the experiments and the bioinformatics analysis. N.B., A.G., J.C.M., and M.K. analyzed the data. N.B., J.C.M., and M.K. wrote the manuscript. All authors approved the final version of the manuscript.
Author Disclosure Statement
No competing financial interests exist.