
Abstract
West Nile virus (WNV) is an emerging arbovirus, circulating worldwide between birds and mosquitoes, which impacts human and animal health. Since the mid-1990s, WNV outbreaks have emerged in Europe and America and represent currently the primary cause of encephalitis in the United States. WNV exhibits a great genetic diversity with at least eight different lineages circulating in the world, and four (1, 2, Koutango, and putative new) are present in Africa. These different WNV lineages are not readily differentiated by serology, and thus, rapid molecular tools are required for diagnostic. We developed here real-time RT-PCR assays for detection and genotyping of African WNV lineages. The specificity of the assays was tested using other flaviviruses circulating in Africa. The sensitivity was determined by testing serial 10-fold dilutions of viruses and RNA standards. The assays provided good specificity and sensitivity and the analytical detection limit was 10 copies/reaction. The RT-PCR assays allowed the detection and genotyping of all WNV isolates in culture medium, human serum, and vertebrate tissues, as well as in field and experimental mosquito samples. Comparing the ratios of genome copy number/infectious virion (plaque-forming units), our study finally revealed new insight on the replication of these different WNV lineages in mosquito cells. Our RT-PCR assays are the first ones allowing the genotyping of all WNV African variants, and this may have important applications in surveillance and epidemiology in Africa and also for monitoring of their emergence in Europe and other continents.
Introduction
W
WNV exhibits a great genetic diversity with at least eight different lineages circulating in the world, without known specific biological properties (Mackenzie and Williams 2009, Fall et al. 2014). Among them, four (i.e., 1, 2, Koutango, and a putative new lineage) are present in Africa, particularly in Senegal. Lineage 1 is the only one found worldwide and associated with all major human outbreaks (Murray et al. 2010, Anukumar et al. 2014, Hernández-Triana et al. 2014). Lineage 2 was exclusively present in Africa, but since 2004, this lineage has been reported circulating in Europe with severe cases in humans and birds (Bakonyi et al. 2006, Hernández-Triana et al. 2014). Viruses with high and low neuro-invasiveness phenotype exist in both lineages 1 and 2. Koutango and the putative new lineage are local African lineages, never isolated in humans, birds, or horses (Fall et al. 2014,
The report of lineage 2 in Europe, which was a local African lineage, suggests ongoing exchanges between Africa and Europe, and migratory birds that overwintered in Africa may have introduced lineage 2 into Europe (Bakonyi et al. 2006, Hernández-Triana et al. 2014). A similar migration event has been also observed for USUV (Nikolay et al. 2013).
Interestingly, Senegal is an important stopover point for bird migration between Africa and Europe in the area of the ornithological park Djoudj on the Senegal River. All these observations suggest the potential of the African local lineages (Koutango and the putative new lineage) to spread beyond their expected geographical areas into Europe.
These different WNV lineages are not readily differentiated by serology; therefore, rapid molecular methods are required for detection and genotyping of WNV in suspect cases and potential vectors. This may have important applications not only in surveillance and epidemiology of African WNV lineages in Africa but also for surveillance of their emergence in Europe and other continents. Molecular tools are available for WNV detection (Linke et al. 2007, Zaayman et al. 2009, Faggioni et al. 2014), but the genetic diversity of all African WNV variants has never been taken into account. In this study, we developed rapid molecular tools for a consensus detection of all WNV lineages (WNV consensus assay) and genotyping of the different WNV lineages reported in Africa (lineage-specific assays).
Materials and Methods
Virus
Fifteen WNV and seven other flavivirus isolates were used in this study and are described in Table 1. The virus stocks were prepared by inoculating Aedes pseudoscutellaris (AP61) continuous cell lines for 4 days, and immunofluorescence assay as previously described (Digoutte et al. 1992) was performed to assess the cell infection with WNV. Virus stocks were titrated as previously described, using porcine stable cells (porcine kidney stable cell line, American Type Culture Collection, Manassas, VA) (De Madrid and Porterfield 1969). The cell culture media were used as viral stocks for RNA extraction and RT-PCR.
The years of isolation, origins, sources, and accession numbers or references are mentioned. DENV2 and DENV4 correspond to Dengue virus serotype 2 and 4.
CAR, Central African Republic; WNV, West Nile virus.
Primer and probe design for WNV consensus assay
To develop a locked nucleic acid probe-based real-time RT-PCR, target regions described previously, envelope gene (E-gene) (Lanciotti et al. 2000) and 3′ untranslated region (3′UTR) (Jiménez-Clavero et al. 2006), were considered. The envelope gene target was dismissed as it had been chosen for detection of American WNV type 1 sequences only. Using 78 sequences available from GenBank at the time of design, an amplicon was designed for the 3′UTR target region. Because of too high divergence, the sequences “Rabensburg” (AY765264) and “LEIV-Krnd88-190” (AY277251) were not included in the alignments. These primers were synthesized (TIB Mol-Biol, Berlin, Germany) and tested to identify a pair, which would not amplify any of the other flaviviruses tested (Table 2).
All probes were tagged 6FAM at the 5′ and BBQ at the 3′ end. + preceding a nucleotide indicates LNA nucleotide.
FP, forward primer; LNA, locked nucleic acid; P, probe; RP, reverse primer; Tm, melting temperature; UTR, untranslated region.
Primer and probe design for WNV lineage-specific assays
To develop lineage-specific-real-time RT-PCR assays, the E-gene sequences of strains belonging to each lineage (1, 2, Koutango, and the putative new lineage) were considered. The different primers and probes designed (Table 2) were synthesized (TIB Mol-Biol) and tested.
RNA standard
For the consensus assay, a 1047 nucleotide fragment of the 3′UTR region was amplified using WNVUTR UP 5′-TGCTTCTGTACTTCCACAGAAGAG-3′ and WNVUTR DP 5′-AGATCCTGTGTTCTCGCACC-3′. For the lineage-specific assays, 727-bp fragments of the E-gene were amplified with the primers WNV E-F (5′-CAACTGCCTAGGAATGAGYAACAG-3′) and WNV E-R (5′-GGCATGAGGTTCTTCAAACTC CA-3′). The obtained PCR products were ligated into pCRII (3′UTR) and pCR2.1 (E-gene) (Life Technologies, GmbH, Darmstadt, Germany) and used for in vitro transcription of an RNA standard with T7 RNA-polymerase (Roche, Mannheim, Germany) as described previously (Nikolay et al. 2014).
Determination of specificity
The specificity of the different assays was determined by testing 10 WNV strains belonging to the 4 lineages circulating in Africa (i.e., 3 lineage-1, 4 lineage-2, 2 Koutango, and the unique strain of the putative new lineage) in comparison to other flavivirus isolates of Dengue virus, USUV, Yellow fever virus, Zika virus, and Bagaza. Before WNV RT-PCR assays, all these isolates were tested using Pan-Flavi assay (Patel et al. 2013) and existing specific assays for Dengue virus (Wagner et al. 2004), Yellow fever virus (Weidmann et al. 2010), and Zika (Faye et al. 2013) as described previously for amplification control.
Determination of sensitivity
Dilutions (10-fold) of each in vitro RNA standard with known initial copy number were quantified in triplicate using the different assays. Regression curves were obtained representing the RNA copy number/reaction versus the threshold cycle (Ct) value. The lowest RNA copy number with RT-PCR detection was considered as the analytical detection limit.
The sensitivity of viral RNA detection was also evaluated by serial 10-fold dilutions of viral stocks with known titer of each WNV type in L-15 medium (Gibco BRL, GrandIsland, NY) and in human serum (Sigma-Aldrich, Saint-Louis, MO). To verify for the number of genome copies per plaque-forming units (pfu), all supernatants were quantified using the respective WNV real-time RT-PCR assays. The WNV reference strain Eg101 was used for lineage 1-specific and consensus RT-PCR assays. The WNV reference strains B956, ArD96655, and ArD94343 were used, respectively, for lineage 2, Koutango, and new lineage-specific assays. The assays were performed in triplicate. For the 10-fold dilutions, regression curves were obtained representing the pfu/reaction versus the Ct value. The lowest titer with RT-PCR detection was considered as the analytical detection limit.
Processing of field samples and experimentally infected mosquitoes
Field mosquito pools, rodents, and experimentally infected mosquitoes (Fall et al. 2014) were used to test the performance of the assays. Experimentally infected mosquitoes were homogenized as previously described (Fall et al. 2014), and mosquito pools collected in the field were homogenized in 3 mL of L-15 supplemented with 20% of fetal bovine serum (FBS). Rodent tissues were homogenized in 3 to 4 mL of L-15 containing 5% FBS. Viral RNA was extracted from the homogenates and analyzed by real-time RT-PCR assays (see RNA extraction and RT-PCR section).
RNA extraction and RT-PCR
RNA was extracted from 100 μL of each viral stock, mosquito or rodent homogenates, and 10-fold dilution of WNV lineage stocks in L-15 medium or human serum using the QIAamp viral RNA extraction kit (Qiagen, Heiden, Germany) according to the manufacturer's instructions.
The real-time RT-PCR assay was performed using an ABI 7500 cycler (Applied Biosystems, Foster City, CA) and the QuantiTect Probe RT-PCR kit (Qiagen). Reactions were performed in 25 μL reaction volume containing 1 μL diluted RNA, 0.5 μM forward and reverse primer, 0.2 μM of probe, 12.5 μL of 2× QuantiTect Probe RT-PCR Master Mix, and 0.25 μL of QuantiTect RT Mix. The RT-PCR conditions were as follows: 15 min 50°C, 10 min 95°C, 40 cycles of 15 s 95°C, and 1 min 60°C.
Determination of intra-assay, interassay reproducibility and efficiency
Samples were extracted and amplified 10 times in the same run to evaluate intra-assay variability and in 10 different runs to evaluate interassay variability. The amplification efficiency of the primers was calculated from the slope of the standard regression lines (E = 101/slope − 1).
Results
Analytical specificity and analytical sensitivity
RNAs from the 10 WNV and other flavivirus isolates were tested using Pan-Flavi, WNV consensus, and WNV lineage-specific real-time RT-PCR assays developed herein. The Pan-Flavi assay detected all the isolates tested. The WNV consensus assay detected all the 10 WNV strains, but initially also detected RNAs from USUV strains (data not shown). Using the amplification refractory mutation system (ARMS) principle (Newton et al. 1989, Weidmann et al. 2003), the reverse primer was adapted to an insertion in the USUV sequences matching the second position of a newly designed WNV reverse primer (Fig. 1). The new WNV consensus assay (Table 2) now did not show any more cross detection of RNAs from USUV strains.

Sequence alignment of 3′UTR sequences of WNV and USUV as indicated by accession numbers with WNV consensus assay primers and LNA probe. Nucleotide positions refer to the sequence of reference strain Eg101 from Egypt (AF260968). AF196835 represents sequences from the United States. AY277252 represents sequences from Russia. Degenerated positions are in bold. LNA, locked nucleic acid; USUV, Usutu virus; UTR, untranslated region; WNV, West Nile virus.
To boost differentiation and therefore specificity of the individual WNV lineage-specific assays, primers were also designed using the ARMS principle with specific matches at the 2 to 3 position of the 3′-end of the primers, mismatching to as many of the other strains as possible (Fig. 2). These lineage-specific assays detected and correctly genotyped the respective WNV strains tested in each lineage, but neither detected other flaviviruses tested nor cross-reacted within other WNV lineages tested (Table 3).
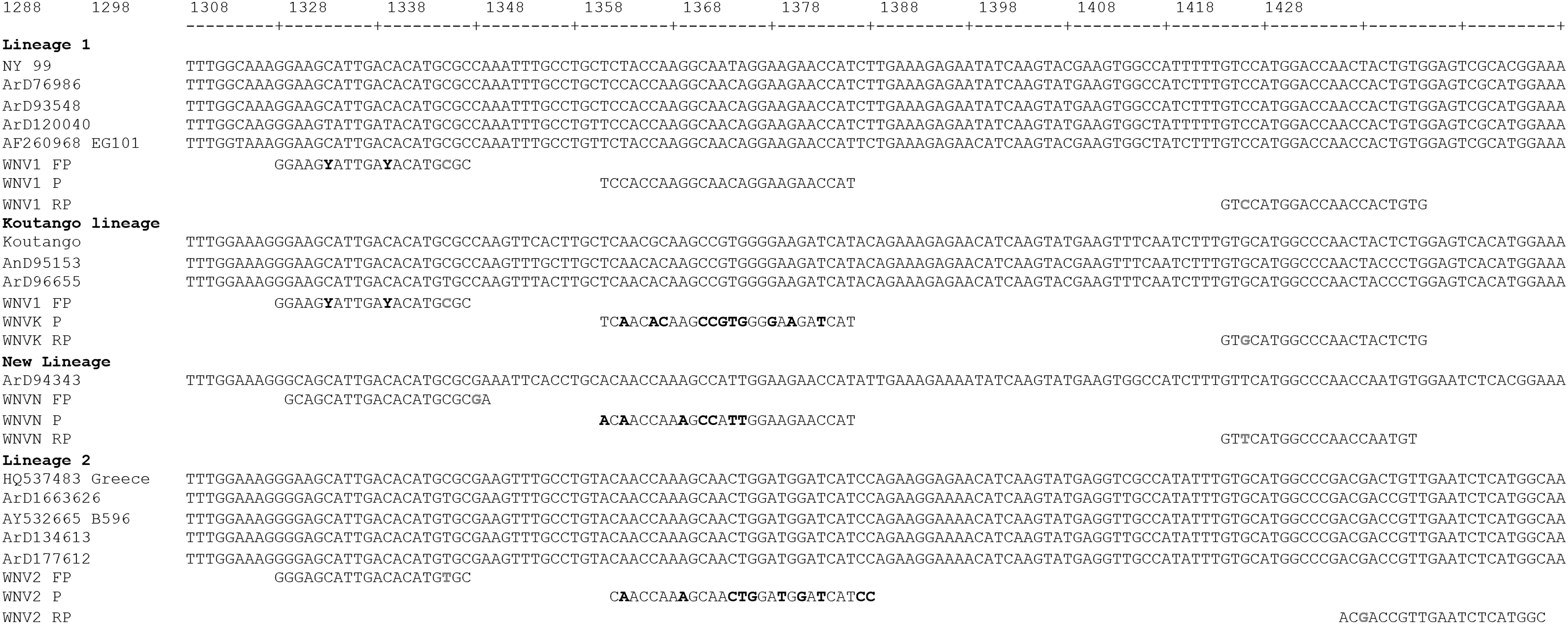
Sequence alignment of WNV E-gene sequences from Africa, Senegal, Egypt, Uganda, Europe, and America as indicated by strain designations with WNV lineage-specific primers and probes. The forward primer WNV FP is common to lineages 1, Koutango, and the European WNV2 isolates. Nucleotide positions refer to the sequence of reference strain Eg101 from Egypt (AF260968). Probe mismatches are highlighted in bold in reference to probe WNV1 P for the detection of WNV lineage 1. Mismatches were introduced to the 3′ ends of the primers according to the ARMS principle to enhance differentiation. ARMS, amplification refractory mutation system; FP, forward primer; P, probe; RP, reverse primer.
Samples were tested by real-time RT-PCR using Pan-Flavi, lineage-specific, and consensus assays in two independent experiments (assay 1 and 2). The Ct (threshold cycle) values are represented.
—, negative amplification.
The performance of the assays was tested by using 10-fold dilutions of in vitro RNA standards. Five independent runs were done using the RNA standards of the consensus and each of the lineage-specific assays (Fig. 3). The analytical detection limit was 10 copies/reaction for all the RT-PCR assays (Fig. 3). The linear regression showed that the different curves were linear with correlation coefficients ≥0.998 in all cases. Efficiencies ranged from 99% to 141%, with lower efficiencies for Koutango (103%) and putative new lineage (99%) assays and higher efficiency for the consensus assay (141%). The lineage 1 and 2 assays were intermediate with 129% and 121%, respectively (Table 4).
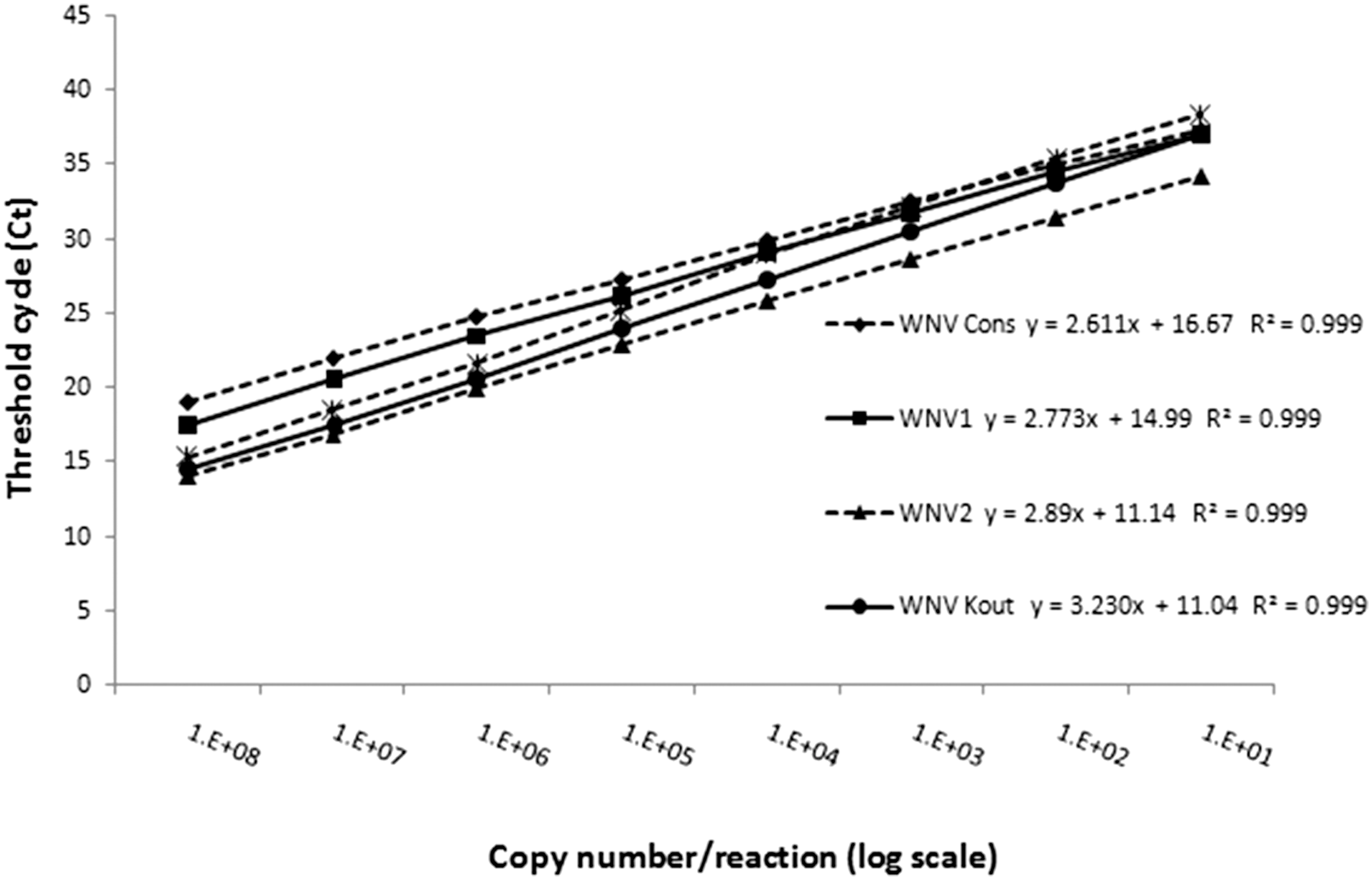
Serial 10-fold dilutions of in vitro RNA standard have been tested in the corresponding real-time RT-PCR assay. Tested dilutions ranged from 1 × 108 to 0.1 copies/reaction for the RNA standard of lineages 1, 2, Koutango, and new lineage-specific assays and consensus assay.
The WNV stocks were quantified using plaque titration and real-time RT-PCR assays. The ratios copy number/pfu and RT-PCR efficiencies were calculated.
pfu, plaque-forming units.
Viral RNA quantification
The determined titers of Eg101, B956, ArD96655, and ArD94343 (corresponding, respectively, to lineages 1, 2, Koutango, and new lineage viral stocks) are presented in Table 4. The RT-PCR analyses on these viral stocks were done using lineage-specific assays, and the copy number and ratio copy number/pfu are presented in Table 4.
Dilutions (10-fold) of Eg101, B956, ArD96655, and ArD94343 prepared in L-15 medium and human serum were analyzed by RT-PCR with the lineage-specific assays. The consensus assay was tested only on strain Eg101 (lineage 1) and yielded a detection limit of 0.13 pfu/reaction (Fig. 4A). The lineage-specific reactions yielded a detection limit of 1.3, 5, 4.49, and 9.16 pfu/reaction, respectively, for the detection of lineage 1, lineage 2, Koutango, and the putative new lineage (Fig. 4B–E).
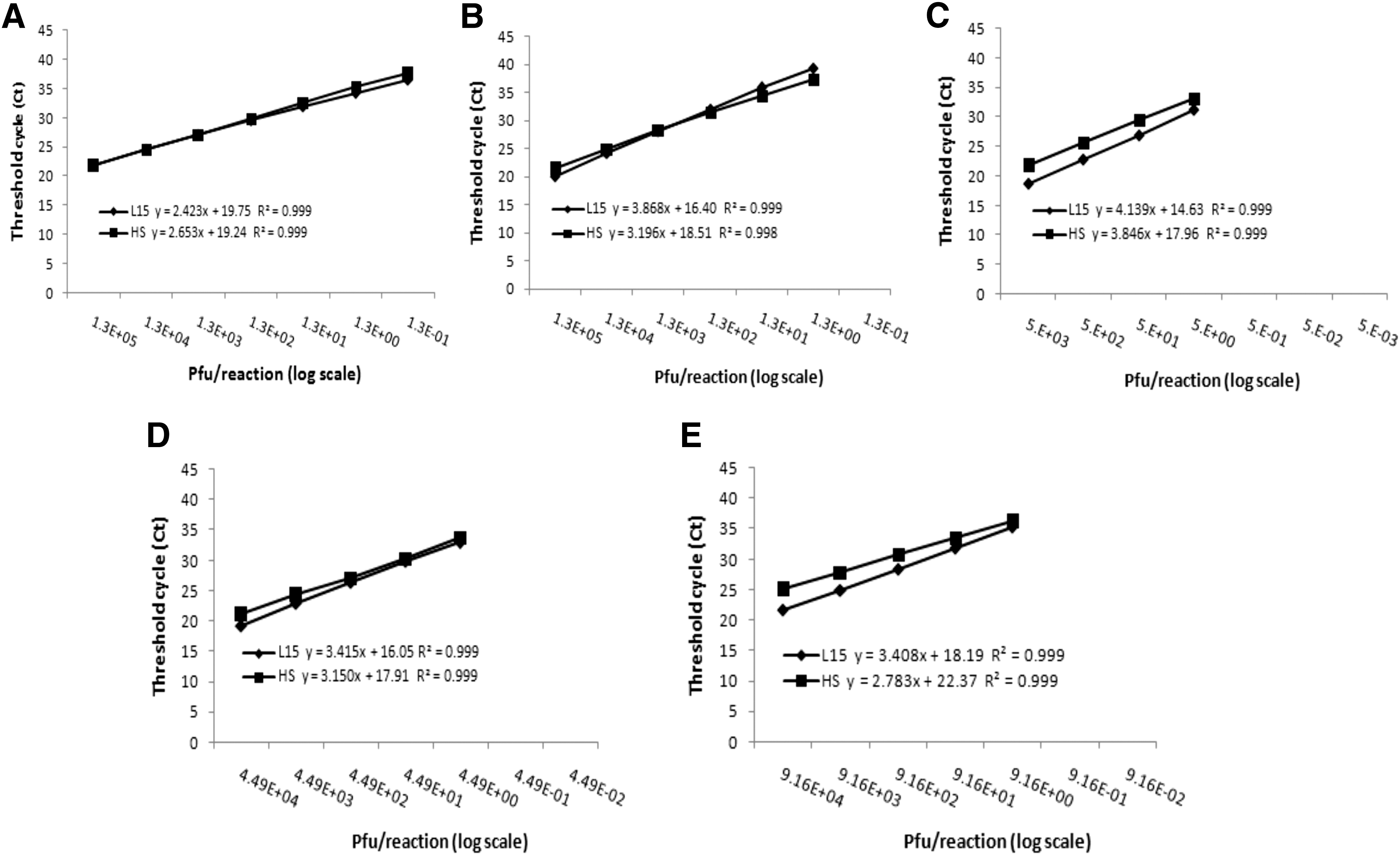
Serial 10-fold dilutions of virus in L-15 medium and human serum have been tested for consensus
The curves obtained were comparable between L-15 and human serum and linear with correlation coefficients ≥0.998. Efficiencies ranged from 74% to 158% for dilutions in L-15, and 82% to 138% for dilutions in human serum, with higher efficiencies for the consensus assay and lower efficiencies for lineage 2-specific assay in both L-15 and human serum.
Processing of field samples and experimentally infected mosquitoes
To assess the performance of our assays, a panel of samples from experimentally infected mosquitoes, wild-caught mosquito pools, and rodent tissues were used. For each experimental sample, the corresponding virus used to infect the mosquitoes was correctly detected and genotyped (Table 5). The field samples naturally infected with WNV lineage 1 or Koutango, as previously assessed by classical RT-PCR and sequencing (data not shown), were correctly detected and genotyped with the lineage-specific assays in the mosquito pools or rodent tissues, including brain homogenates (Table 5).
The Ct (threshold cycle) values are represented.
Field viruses were isolated in 2012 and 2013 in Senegal, and experimental viruses are presented in Table 1. Samples were tested by real-time RT-PCR using lineage-specific assays in two independent experiments (assays 1 and 2).
—, negative amplification.
Variability intra- and interassay
The intra-assay coefficients of variation (CV) ranged from 1.31% to 1.75% and the interassay CV ranged from 2.66% to 4.62% for all assays.
Discussion
Molecular techniques are faster, more accurate, and sensitive for virus detection compared to culture methods. In this study, we established molecular tools for a consensus detection of all WNV lineages and genotyping of the lineages reported in Africa. The good sensitivity of the assays allows the detection and genotyping of all WNV isolates in culture medium, human serum, as well as in field and experimental samples. After the development of the lineage-specific assays, new WNV lineage 2 sequences described in Europe became available (Barzon et al. 2013). Since they introduce a mismatch at position 3 from the 3′-end of the lineage 2-specific forward primer, we suggest using the lineage 1-specific upstream primer, which is also used for Koutango for highly sensitive detection of these strains (Fig. 2). However, we were not able to test these strains in the frame of this study.
Our assays allowed the quantification of WNV RNA by using quantitative RNA standards, yielding viral loads in mosquito and vertebrate samples.
Using the RNA standard, the different assays were very sensitive and able to detect 10 RNA molecules. Our RT-PCR assays therefore exhibit analytical sensitivity similar to others using WNV lineages 1 and 2 (Jiménez-Clavero et al. 2006, Barros et al. 2013, Del Amo et al. 2013).
Using viral RNA, the consensus and lineage 1-specific assay were more sensitive and were able to detect less than 2 pfu of virus. Using the same strain for the consensus and lineage 1-specific assays showed that the consensus assay is more sensitive for detection of low viral loads. This is reflected by the different ratios copy number/pfu obtained for Eg101 (56.08 with the consensus assay and 34.34 with the lineage 1-specific assay) and can be correlated with the higher efficiencies of the consensus RT-PCR assay.
Our results also showed that the different WNV lineages can be detected in human serum with slightly higher efficiencies, compared to culture medium, and in vertebrate tissues, enabling the early and efficient diagnosis of these different WNV lineages from clinical samples. Finally, our study revealed a new insight on the replication of these particular WNV strains in AP61 mosquito cells. The ratios genome copy number/infectious virion (pfu) showed that the new lineage strain and Koutango replication were very efficient with few genomes per pfu indicating apparently a high rate of infectious particles produced, while lineage 2 appeared to overproduce genomes indicating less efficient packaging and possibly more defective particles during its replication (Weidmann et al. 2011).
Conclusions
To our knowledge, the RT-PCR assays described in this article are the first ones that allow the detection and genotyping of all reported African WNV variants.
Surveillance programs now can target all WNV variants, and our tools can be used to monitor the prevalence of WNV virus lineages in mosquito vectors and vertebrates and may be applied in diagnosis as well as in epidemiology and surveillance programs. This is particularly relevant for the surveillance of WNV lineages emerging in geographic regions where they have not previously been identified. The consensus assay is the most sensitive in all tested materials and does not cross detect any of the tested flaviviruses and therefore is an ideal diagnostic assay covering the breadth of WNV strains worldwide.
Therefore, our tools are efficient for rapid detection of African WNV lineages, however, they cannot replace conventional techniques (serology, virus isolation, or molecular sequencing), which are still needed for in-depth characterization of the virus.
Footnotes
Acknowledgments
The authors thank Moussa Dia, Mireille Mondo, Arame Ba, and Khardiata Mbaye for their excellent technical assistance. This work has been financially supported by Institut Pasteur de Dakar and funded by EU grant HEALTH.2010.2.3.3-3-261391 EuroWestNile.
Author Disclosure Statement
No competing financial interests exist.