
Abstract
Voles (Arvicolinae, Rodentia) are known carriers of zoonotic bacteria such as Bartonella spp. and Francisella tularensis. However, apart from F. tularensis, the bacterial microbiome of voles has not previously been determined in Finland and rarely elsewhere. Therefore, we studied liver samples from 61 voles using 16S ribosomal RNA gene PCR analysis, followed by Sanger sequencing. Twenty-three of these samples were also studied with tag-encoded pyrosequencing. The samples originated from 21 field voles (Microtus agrestis), 37 tundra voles (Microtus oeconomus), and 3 bank voles (Myodes glareolus). With the more conventional 16S rDNA PCR analysis, 90 (33%) of the recovered 269 sequence types could be identified to genus level, including Bartonella, Francisella, Mycoplasma, Anaplasma, and Acinetobacter in 31, 15, 9, 9, and 9 sequences, respectively. Seventy-five (28%) matched best with sequences of uncultured bacteria, of which 40/75 could be classified to the order Clostridiales and, more specifically, to families Lachnospiraceae and Ruminococcaceae. Pyrosequencing from 23 samples revealed comparable and similar results: clinically relevant bacterial families such as Mycoplasmataceae, Bartonellaceae, Anaplasmataceae, and Francisellaceae were recognized. These analyses revealed significant bacterial diversity in vole livers, consisting of distinct and constant sequence patterns reflecting bacteria found in the intestinal gut, but including some known zoonotic pathogens as well. The molecular bacterial sequence types determined with the two different techniques shared major similarities and verified remarkable congruency between the methods.
Introduction
V
16S rRNA gene sequencing has provided a strong alternative to traditional culture-based identification of bacteria (Weisburg et al. 1991) in clinical microbiology (Salipante et al. 2013) and medicine (Winglee et al. 2014), as well as in extensive projects such as the Human Microbiome Project (Human Microbiome Project Consortium 2012).
Broad-range 16S rDNA PCR (Br-PCR) is based on universal primers detecting conserved regions in the chromosome coding 16S rRNA genes (Schmidt and Relman 1994), followed by bacterial identification by Sanger sequencing of intervening variable and hypervariable DNA regions. Alternatively, 16S rRNA gene sequences can be retrieved by using tag-encoded primers targeting hypervariable regions, followed by the use of next-generation sequencing technology (e.g., pyrosequencing) that generates hundreds of thousands of sequences in a single run (Shendure and Ji 2008). Both techniques enable not only identification of previously known bacterial species but also discovery of DNA sequences of previously uncharacterized bacteria. Recently, microbial populations have been studied using next-generation techniques to investigate, for example, ticks in Japan (Qiu et al. 2014) and voles in France (Razzauti et al. 2015).
The main objective of this work was to determine the vole microbiome present in livers and to identify potential zoonotic pathogens of vole communities in Finland. Other objectives were to compare the two screening methods, conventional Br-PCR and tag pyrosequencing, and to evaluate their suitability for investigating complex, polymicrobial bacterial DNA samples.
Materials and Methods
Sample collection
Altogether, 60 snap-trapped voles with splenomegaly originating from three locations in Finnish Lapland (Kolari, Pisavaara, and Pyhätunturi) and two locations in Central Finland (Konnevesi and Pieksämäki) were studied. Altogether, 21 field voles (Microtus agrestis), 36 tundra voles (Microtus oeconomus), and three bank voles (Myodes glareolus) were included (Table 1). In addition, one tundra vole from Lapland without splenomegaly served as a control. Three voles from Central Finland had been previously identified positive for F. tularensis by PCR (Rossow et al. 2014c). Liver was chosen as the optimal organ for screening based on our earlier studies (Rossow et al. 2014c).
Three voles highly positive for Francisella tularensis.
Located in northern Finland.
No splenomegaly in one vole.
Traps were set in the evening and collected soon after sunrise. During the trappings, either in September in Lapland or early May or October in Central Finland, temperatures remained between 0°C and 10°C. Therefore, voles caught in the morning were fresh. They were dissected or frozen immediately.
Ethics statement
No ethical permit was needed because the Finnish Act on the Use of Animals for Experimental Purposes (62/2006) and the Finnish Animal Experiment Board (16th May, 2007) do not classify snap-trapping as an animal experiment. However, a permit (23/5713/2001) for capturing protected species (as bycatch) was granted by the Finnish Ministry of the Environment; none of our target species belonged to the protected ones.
DNA extraction
DNA was extracted from about 20 mg of liver tissue with the Wizard Genomic DNA Purification Kit (Promega). Additionally, three negative controls containing nuclease-free water (W4502; Sigma-Aldrich) instead of sample were used to screen for bacterial DNA contamination in extraction reagents.
Conventional Br-PCR analysis
DNA from 61 vole samples was used for amplification of a partial bacterial 16S rRNA gene sequence (∼550 bp, 8F-575R) with universal primers, fD1mod (Kotilainen et al. 1998) and 16S1RR-B (Wilbrink et al. 1998). To assess potential contaminant sequences from extraction and PCR reagents, two different contamination libraries were established: First, for investigating exogenous bacterial DNA in extraction reagents, three negative extraction controls were analyzed. Second, to study the presence of contaminating DNA in PCR reagents, nuclease-free water was used as template in no template control (NTC) reactions, utilizing two different reaction mixtures. To avoid false-positive results in PCR, careful preventive measures were taken (Kwok and Higuchi 1989, Lo and Chan 2006). (Materials and Methods section in the Supplementary Data; Supplementary Data are available online at
Br-PCR amplification reaction mixture in the volume of 50 μL contained Gene Amp® PCR Buffer (Life Technologies [LT]), 200 μM of each dNTP (LT), 0.4 μM of each primer, 4 mM MgCl2 (LT), 2 U AmpliTaq Gold® DNA Polymerase LD (LT), 5 μL template, and nuclease-free water (W4502; Sigma-Aldrich). PCRs were performed using the DNA Engine (PTC-200) Peltier Thermal Cycler (Bio-Rad Life Sciences). Reaction conditions were 3 min at 94°C, followed by 30 cycles of amplification at 94°C for 30 s, 60°C for 30 s, and 72°C for 30 s, and the final extension step at 72°C for 10 min.
PCR products of the correct size were cloned to Escherichia coli vectors using the TOPO TA Cloning Kit according to the manufacturer's instructions (Invitrogen Corporation). Randomly selected individual colonies were amplified directly by PCR using the M13 priming sites. Amplicons of the expected size were sequenced using Applied Biosystems Dye Terminator (v.3.1) sequencing kit (LT) and reactions were run on 3100xl Capillary Sequence Analyzer (LT) at the University of Helsinki (Sequencing Core Facility). Sequences were edited with the Sequencher 5.1 program (Gene Codes Corporation).
In addition to BLAST analysis (
Tag pyrosequencing
Liver DNA samples from 22 voles with and one without splenomegaly were used for tag-encoded pyrosequencing analysis of the 16S rRNA gene (V1-V3 region, 27F-518R), as described previously (Hanski et al. 2012), except 35–45 cycles were used in PCR runs. Additionally, three negative extraction controls pooled together were studied. Sequencing of PCR products using the 454 platform was done at Institute of Biotechnology, University of Helsinki (Finland). The sample-specific sequences were uploaded into the RDP Classifier (release 11.2) (Wang et al. 2007) to identify the bacterial classes and genera, with 80% as the threshold value. Sequences present in the pooled negative extraction control were omitted from the final results. Samples were considered positive for a specific bacterial sequence type when the number of sequence reads exceeded 10.
Further sequence data analyzing was done using mothur (Schloss et al. 2009) according to workflow described by Schloss et al. (2011). Before analyzing the sequences, the tag, primer sequences, and low-quality sequences (i.e., ambiguous nucleotides, homopolymers longer than eight nucleotides, average quality score less than 35) were removed. Sequences were aligned to Greengenes reference database (DeSantis et al. 2006); chimeras were detected using UCHIME algorithm (Edgar et al. 2011). Chimeric sequences and sequences present in the pooled negative extraction control were removed from the dataset. Cutoff of 0.03 was used for the clustering of operational taxonomic units (OTUs). Alpha diversity was estimated using Shannon and inverse Simpson indexes. The Yue and Clayton measure of dissimilarity was used for dendrogram construction and for the Principal Coordinate Analysis (PCoA).
Results
With conventional Br-PCR, 408 randomly selected clone sequences from the 61 vole liver samples were sequenced. Fourteen sequences of poor quality were excluded from the final results. In addition, one plastid sequence and one sequence deciphered as a chimera were removed from the final results. Furthermore, 12 and 96 of the sequences represented the host species, bank vole and field vole, respectively (Supplementary Table S1). These sequences and a total of 15 sequences that shared over 99% similarity with sequences from the contaminant libraries were omitted from the final results. Of the remaining 269 sequence types, 33% (90/269) were identifiable at least to genus level, including Bartonella, Francisella, Mycoplasma, Anaplasma (including Ehrlichia phagocytophila) (Dumler et al. 2001), and Acinetobacter in 31, 15, 9, 9, and 9 sequences, respectively (Table 2). Furthermore, 28% (75/269) matched best with uncultured bacteria. Sequence analysis with the RDP Classifier revealed that over half (40/75) of those were classified to the order Clostridiales and, more specifically, to families Lachnospiraceae and Ruminococcaceae (Supplementary Table S1). Interestingly, a noteworthy group of sequences comprising 33% (89/269) of all the data shared 89–93% similarity with Mycoplasma spp. and could only be classified as bacteria using RDP.
Sequences derived from voles highly positive for F. tularensis (Rossow et al. 2014c).
Six sequences derived from the control vole without splenomegaly: two uncultured Firmicutes bacteria, two uncultured bacteria, two closest match: uncultured bacteria (94–95%).
Percentage of sequence similarity to previously published sequences is indicated in brackets, given only for values <97%.
Altogether, 23 vole liver-derived DNA samples and a pooled extraction control sample were also analyzed using 16S rRNA gene pyrosequencing. Contaminant sequence types present in the pooled extraction control were omitted from the final results (Supplementary Table S2). Majority of the remaining reads represented Francisellaceae (28%) and Bartonellaceae (17%) (Fig. 1). Other common families were Anaplasmataceae (9%), Lachnospiraceae (7%), Ruminococcaceae (6%), Porphyromonadaceae (6%), and Mycoplasmataceae (4%). In addition, 11% of the sequence reads belonged to an artificial unclassified taxon comprising unidentifiable sequences. The amount of sequence reads obtained varied from 3580 reads to over 24 000 reads per sample (Fig. 2a). The relevant sequence types detected from the vole samples are presented in Figure 2b. In general, Br-PCR and pyrosequencing gave comparable results. This can be seen in Table 3, where the most common findings are compared. In 17 samples of 23, the most abundant findings were in congruence.
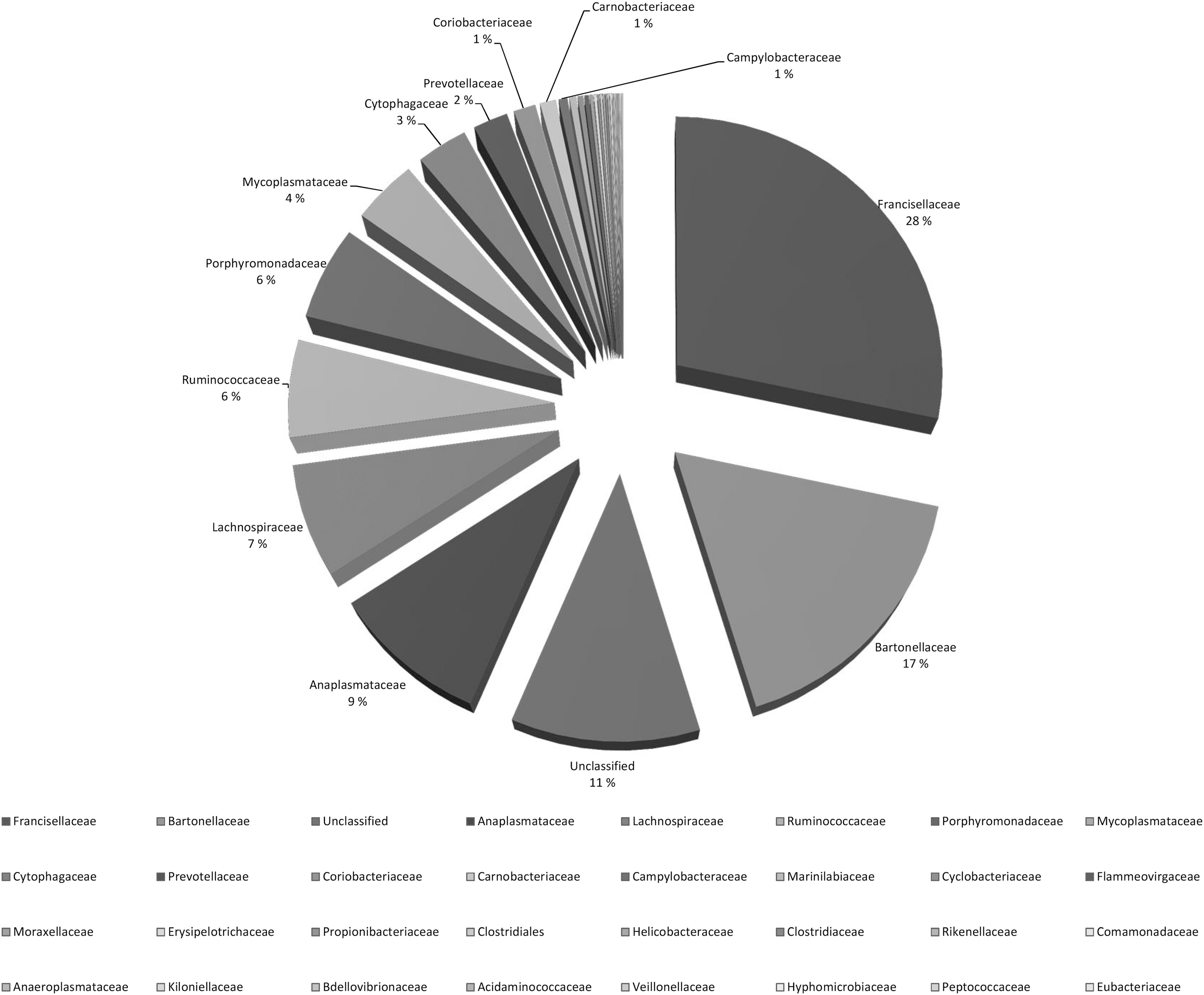
Relative percentage of bacterial families from 23 vole DNA samples based on the number of sequence reads with pyrosequencing. Contaminant sequences (Halomonas, Shewanella, and contaminant sequences based on our negative extraction control) and sequences in which the total number of reads per bacterial family was fewer than one hundred are omitted. Only bacterial families with read percentages >1% are indicated.
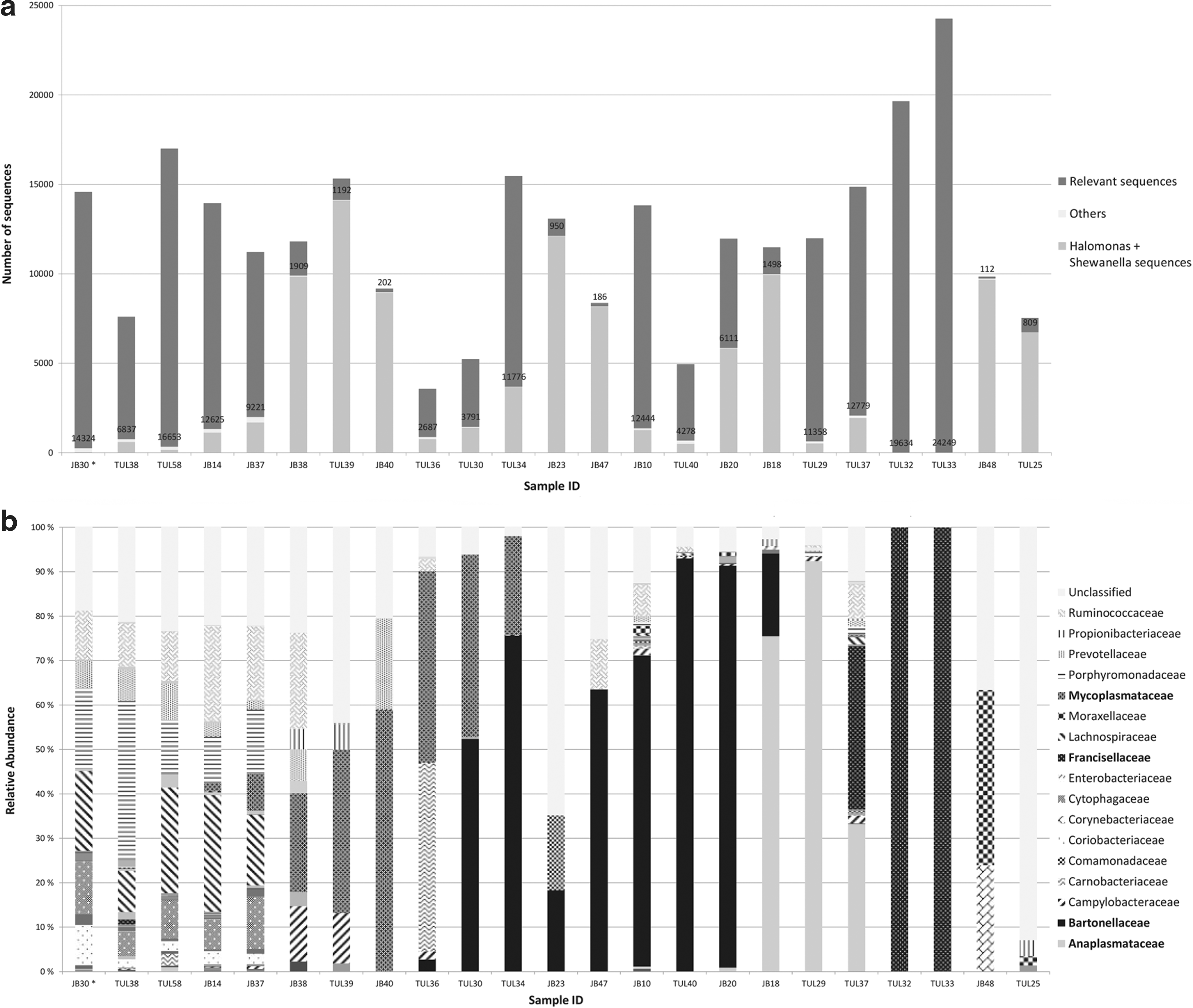
Sequence types detected from vole liver tissue with pyrosequencing.
Includes findings with assignment confidence of ≥97%.
Includes findings of sequence types, which exceed the relative abundance of 5% in the sample. Percentage is indicated in brackets.
Control vole; no splenomegaly.
Results with BLAST search tool: Mycoplasma sp. (93%).
Using conventional Br-PCR, 27 cloned sequences from three negative extraction controls were obtained and these represented 11 sequence types (Supplementary Table S3). BLAST analysis revealed that 12 of the sequence types had 100% identity with Propionibacterium acnes. To determine DNA remnants present in PCR reagents, altogether 529 sequences from 19 NTCs were analyzed using conventional Br-PCR. Sequences were cloned and sequenced also from NTCs from which no amplification product was seen in the agarose gel (Supplementary Fig. S1). The main contaminant sequence types identified were Lactococcus lactis, uncultured Burkholderiaceae bacterium, Phyllobacterium sp., Schlegelella sp./Leptotrix sp., uncultured Sulfurospirillum sp., uncultured Ruminococcaceae bacterium, Altererythrobacter sp., Propionibacterium sp., and Clostridium sp. (Supplementary Table S4). With 454 pyrosequencing, the main bacterial findings in the pooled negative extraction control belonged to genera Halomonas, Shewanella, Pseudomonas, Citrobacter, Bacteroides, and Enterococcus (Supplementary Table S2).
Shannon and inverse Simpson diversity indexes for each sample are shown in Table 4. There were no statistically significant differences between bacterial diversities of different vole species. Heatmap of the relative abundance of bacterial taxons is shown in Supplementary Figure S2. No clear-cut differences between bacterial communities of M. agrestis and M. oeconomus were observed in the heatmap.
Sobs, number of observed OTUs; Inv Simpson, Inverse Simpson's diversity index, with lower (lci) and upper (hci) 95% confidence intervals; Shannon, Shannon index, with lower (lci) and upper (hci) 95% confidence intervals.
The Yue and Clayton measure of dissimilarity between the bacterial community structures was estimated and the resulting distance matrix was used for dendrogram construction (Supplementary Fig. S3) and for the PCoA. While parsimony and unweighted UniFrac methods suggested no statistically significant clustering in respect to vole species or the geographic region of the sampled voles, the weighted UniFrac method (that takes the branch lengths of the dendrogram into account) suggested differences between the clustering (p < 0.001).
PCoA plot is shown in Figure 3. Analysis of molecular variance (AMOVA) was conducted to determine if the spatial separation in the PCoA plot was statistically significant, that is, voles of the same species or voles sampled from the same location shared similar communities and similar abundance levels. There were no statistically significant differences between the bacterial communities of different vole species. However, AMOVA suggested differences when comparing voles captured in Pisavaara and Kolari.
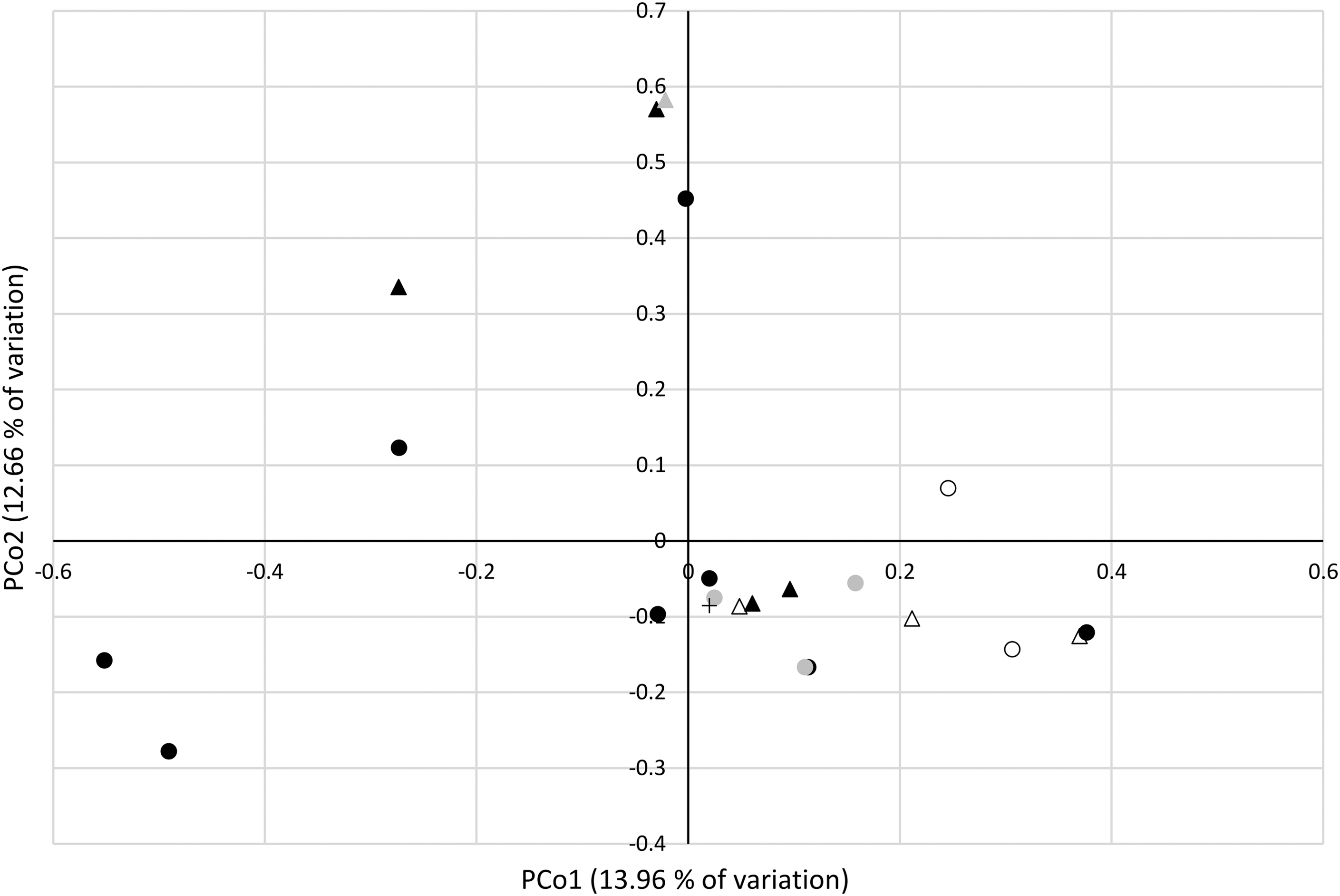
Principal Coordinates Analysis (PCoA). Microtus agrestis samples are indicated with circles, Microtus oeconomus with triangles and M. oeconomus control vole with a cross. The collection sites are indicated as follows: Kolari empty circle/triangle; Konnevesi black circle; Pisavaara black triangle; Pieksamäki gray circle; Pyhätunturi gray triangle. Note that the two principle coordinated explain only 26.62% of the total variation.
Finally, nonparametric T-test (White et al. 2009) was used to determine whether there are differently represented OTUs between vole species or between M. oeconomus voles captured in Pisavaara and Kolari. Between the vole species, this analysis suggested differences in the abundance of Francisella (p = 0.0439), unclassified Firmicutes (p = 0.0409), Clostridiales (p = 0.0010), and Lachnospiraceae (p = 0.0048). Between M. oeconomus voles captured in Pisavaara and Kolari, the analysis suggested different abundance levels for Oscillibacter (p = 0.0343) and Mycoplasma (p = 0.0060).
Discussion
We identified bacterial sequence types in vole liver samples using two metagenomic techniques targeting 16S rRNA genes: conventional Br-PCR and tag pyrosequencing. We also compared the two methods and studied their suitability for investigating complex, polymicrobial bacterial DNA samples.
Sixty-one vole samples were investigated using the more conventional Br-PCR analysis. The main clinically relevant bacterial sequences detected belonged to the genera Bartonella, Francisella, Mycoplasma, Anaplasma, and Acinetobacter. Based on these findings, 23 samples were selected for pyrosequencing. These included one sample from the control vole without splenomegaly and three samples previously identified positive for F. tularensis (Rossow et al. 2014c), as well as an extraction control.
Using pyrosequencing, the bacterial composition of four samples lacked any clinically relevant pathogens, containing mainly members of the families Ruminococcaceae, Porphyromonadaceae, and Lachnospiraceae, in addition to an artificial unclassified taxon (Fig. 2b and Supplementary Table S1). This bacterial pattern was very similar to the control vole's microflora and was perceived as intestinal normal flora. Sixteen samples, however, had a completely distinctive bacterial profile, with families such as Mycoplasmataceae, Bartonellaceae, Anaplasmataceae, or Francisellaceae being highly predominant. In the two remaining samples, Bartonellaceae was present in one sample according to the conventional Br-PCR, but pyrosequencing revealed mainly sequence types belonging to Corynebacteriaceae and Moraxellaceae families. In addition, vole TUL25, which had sequence types sharing 89–93% similarity with Mycoplasma spp., with conventional Br-PCR, gave an unclear bacterial profile with pyrosequencing and hence almost 90% of the reads belonged to the unclassified group. The results of three Francisella-positive samples corresponded with the findings of Rossow et al. (2014c): sequences of the highly positive voles, TUL32 and TUL33, were dominated by Francisellaceae, whereas one vole (TUL37), exhibiting lower positivity in F. tularensis-specific PCR, also contained, for example, Anaplasmataceae besides Francisellaceae. Overall, pyrosequencing analysis of 21 liver samples gave congruent results to those of the Br-PCR (Table 3).
Mycoplasma spp., more specifically M. moatsii- and rarely M. penetrans-specific sequence types, were found moderately often in voles from both Central Finland and Lapland. Mycoplasma infection is a leading cause of pneumonia worldwide in humans (Ishiguro et al. 2013) and other species, including laboratory rodents (Lindsey and Cassell 1973, Sun et al. 2013). Wild rodents commonly carry Mycoplasma spp. (Sashida et al. 2013, Sumithra et al. 2013), but reports on clinical disease are almost nonexistent. M. moatsii may colonize intestines of wild rats (Giebel et al. 1990). More detailed studies on Mycoplasma in wild rodents are certainly needed.
Bartonella spp. were commonly seen in the voles. The animal reservoir for bartonellae is large, including rodents such as rats, mice, and voles (Krauss et al. 2003, Buffet et al. 2012, 2013, Hayman et al. 2013, Silaghi et al. 2016). Human bartonellosis in Finland is rare, but may be life-threatening (Jalava et al. 1995). Our findings included both undetermined (spp.) sequences and sequence types representing known species, such as Bartonella rochalimae, Bartonella taylorii, Bartonella grahamii, and Bartonella henselae. All these have been found in wild rodents (Krauss et al. 2003); the first three (or their close relatives) are common findings in bank and field voles (Buffet et al. 2012, 2013).
Numerous sequences belonged to Anaplasmataceae and Anaplasma. Within this genus, Anaplasma phagocytophilum infections are well known in the bank vole, field vole, common vole (Microtus arvalis), and common shrew (Sorex araneus), but these small mammals harbor a subtype different from those causing tick-transmitted granulocytic anaplasmosis of veterinary and medical importance (Brown et al. 2001, Majazki et al. 2013). A. phagocytophilum is common in bank voles in Finland (Kallio et al. 2014).
Among several other mammals, wild rodents host zoonotic F. tularensis (Rossow et al. 2014b, 2014c), which was present also in this study. Furthermore, DNA belonging to Corynebacteriaceae family was detected from the liver samples. At least two species, Corynebacterium pseudotuberculosis and Corynebacterium ulcerans, colonize or infect ruminants and horses and occasionally humans (Krauss et al. 2003). However, the most notable human infection is caused by C. diphtheria that is considered nearly exclusively as a human pathogen (Sing et al. 2016).
Many of our findings reflect the normal intestinal flora of other rodent species. We found bacterial DNA representing members of the order Clostridiales, some of which (e.g., Clostridium spp.) are well-known parts of normal murine and probably also voles' intestinal flora (Salzman et al. 2002). More specifically, representatives from families Lachnospiraceae and Ruminococcaceae were common. More experimental studies are needed to determine whether our findings reflect nonviable bacterial remnants in the liver or leakage of the intestine walls as part of early decomposition.
Family Moraxellaceae and, more specifically, Acinetobacter were also present in our vole samples. Acinetobacteria are common in nature and occur in normal flora of mice (Pedron et al. 2012) and humans (Bergogne-Berezin et al. 1996). However, every now and then these opportunistic bacteria cause nosocomial epidemics, which may lead to lethal outcomes resulting, for example, from pneumonia. Several fatal infections have also been described in military personnel [reviewed in O'Shea (2012)].
The composition of the intestinal commensal microflora of wild voles is poorly known. However, that of laboratory mice has been studied decades ago by cultivation (Dubos et al. 1965, Schaedler et al. 1965) and later by molecular and microdissection methods (Salzman et al. 2002, Nava et al. 2011, Pedron et al. 2012). According to these studies, murine microflora includes obligate anaerobes, such as Bacteroides spp. and Clostridium spp, facultative anaerobes, such as Lactobacillus spp., Enterococcus spp., and Enterobacillus spp., as well as aerobic Acinetobacter spp. In the mouse colon, bacteria belonging to the phylum Firmicutes are enriched, Lachnospiraceae and Ruminococcaceae being the predominant families (Nava et al. 2011). In addition to Firmicutes, bacteria from phylum Bacteroidetes contribute to the luminal bacterial contents in mice and humans (Ley et al. 2005, Garner et al. 2009, Nava et al. 2011). In murine crypta, Acinetobacter spp. is common (Pedron et al. 2012). These previous findings from mice reflected those seen in our study on voles.
While the OTU-based analysis of the pyrosequencing data suggested some differences between the bacterial communities of different vole species and between some of the locations, these results should be interpreted cautiously. The AMOVA suggested differences between voles captured in Pisavaara compared with those captured in Kolari. This may be due to relatively high proportion of Bartonellaceae in the M. oeconomus voles captured in Pisavaara and the relatively high proportions of Mycoplasmataceae in the voles captured in Kolari. We purposely studied splenomegalic voles as an indicator of potential infection; our results may not represent the average normal flora. The nonpathogenic findings from the sole vole with visibly normal spleen suggest this as well.
Razzauti et al. (2015) studied voles collected from France using next-generation sequencing techniques and detected 45 potential zoonotic bacterial genera from spleen samples. These included, for example, Bartonella, Rickettsia, Borrelia, Neoehrlichia, Anaplasma, and Francisella. Several same bacterial genera were also discovered from our voles. The differences may be due to geographical location, the organs studied, and techniques used, as well as our selection based on splenomegaly.
We found pyrosequencing useful in providing wide-ranging and vast information on the bacterial composition of vole liver tissue. Tagged pyrosequencing is less laborious, resulting in less hands-on time compared with the conventional Br-PCR method as 16S rDNA PCR products can be characterized directly without cloning. Furthermore, since barcoded PCR primers allow dozens of different samples to be analyzed in the same pyrosequencing run, microbial communities can be characterized at a fast rate. Overall, pyrosequencing proved to be a favorable method for characterization of bacterial flora, especially if the number of samples is large. On the other hand, conventional Br-PCR is useful when studying fewer samples and when high-technology instrumentation and expertise are unavailable.
To obtain sufficient amount of PCR product for pyrosequencing, we had to increase the number of PCR cycles from 35 to 45. This raised the amount of contaminant sequences in the final results. In seven samples, for example, the amount of Halomonadaceae and Shewanellaceae reads exceeded even 8000 (Fig. 1a). Previous studies have similarly shown increasing proportion of contaminant sequences and their dominance in samples containing low bacterial copy numbers (Salter et al. 2014). In our study, this was especially evident with three samples, where the relevant sequences numbered only hundreds.
The DECIPHER tool detected one chimera from our Br-PCR data. In addition, it was unable to classify several sequences. According to Wright et al. (2012), indecipherable sequences are often either chimeric or not 16S. Furthermore, altogether 108 sequences shared 78–100% similarity with vole sequences (field and bank vole), thus not being of bacterial 16S rRNA origin (Supplementary Table S1).
We were able to identify 33% of the Br-PCR sequence types to genus level, which leaves a substantial portion of undetermined sequences. These probably represent previously uncharacterized bacteria occurring in vole communities in Finland. Of special interest is a group of sequence types sharing some similarity with Mycoplasma spp. The significance of these findings needs clarification with genus-specific identification methods.
According to this study, contaminating exogenous DNA is common in extraction and PCR reagents and it originates from a variety of bacteria (Supplementary Tables S2–S4). Some of the bacterial species, for example, Pseudomonas, Micrococcus, Sphingomonas, Methylobacterium, Acidovorax, and Phyllobacterium, have been reported earlier (Maiwald et al. 1994, Tanner et al. 1998, Barton et al. 2006), but some novel sequence types were also found. Different PCR reagents and extraction kits seem to contain different contaminants.
Interestingly, many of the contaminant sequence types (such as Phyllobacterium sp., Sphingomonas sp., Variovorax sp., and Pseudomonas sp.) have been reported to exist in several body sites based on a method similar to this study (Supplementary Fig. S4) and published in the Human Microbiome Project (
Conclusions
This study revealed significant bacterial diversity in vole liver samples, consisting of known pathogens and reflecting that of intestinal flora as well. Overall, the findings were independent from the trapping location. The voles commonly carried potential or verified zoonotic bacterial pathogens and therefore they may play a role in spreading them.
The two methods used gave comparable results. Pyrosequencing needs less hands-on time, being more suitable with a large sample number. On the other hand, the Br-PCR method is ideal when studying fewer samples. In addition, our results remind us of the need to acknowledge the possible presence of exogenous DNA in molecular reagents. Ignorance may lead to false conclusions.
Nucleotide sequence accession numbers
The sequences reported in this article were deposited in NCBI GenBank with the accession numbers KT961130–KT961324.
Footnotes
Acknowledgments
The authors thank Heidi Rossow for sample preparation. The authors wish to acknowledge CSC – IT Center for Science, Finland, for computational resources. This work is part of the European Biodefence project EDA B-1325-ESM4-GP and has been partially funded by EU grant FP7-261504 EDENext and cataloged as EDENext 442 (
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.