
Abstract
Getah virus (GETV; genus Alphavirus, family Togaviridae) is a mosquito-borne virus known to cause disease in horses and pigs. In 2014, for the first time in ∼30 years, a sudden GETV outbreak occurred among racehorses in Ibaraki, Japan. Two years before this outbreak, we obtained multiple GETV isolates from Culex tritaeniorhynchus mosquitoes collected in Nagasaki, Japan and determined the whole genome sequence of GETV isolate 12IH26. Our phylogenetic analysis of GETV strains revealed that the isolate 12IH26 forms a robust clade with the epidemic strains 14-I-605-C1 and 14-I-605-C2 isolated from horses in the 2014 outbreak in Ibaraki. Furthermore, the complete genomic sequence of the isolate 12IH26 was 99.9% identical to those of the 2014 epidemic strains in Ibaraki. Phylogenetic analysis also showed that the recent Japanese GETV strains, including the isolate 12IH26, are closely related to the Chinese and South Korean strains rather than the previous Japanese strains, suggesting that GETV strains may be transported from overseas into Japan through long-distance migration of the infected mosquitoes or migratory birds.
Introduction
T
Getah virus (GETV) is a member of alphaviruses, which was first isolated from the mosquito Culex gelidus in Malaysia in 1955 (Berge 1975). Sagiyama virus (SAGV) was isolated from the mosquito Cx. tritaeniorhynchus at nearly the same time (Scherer et al. 1962), but SAGV is now considered to be a strain of GETV (Shirako and Yamaguchi 2000). The serological evidence of GETV infection has been confirmed in various mammals and birds, although GETV is not known to cause disease in numerous animals, including human (Doherty et al. 1966, Simpson et al. 1975, Li et al. 1992). In contrast, GETV infection often causes clinical symptoms, such as fever, urticarial rash, and leg edema in horses (Kamada et al. 1980), as well as fetal death and reproductive disorders in domestic pigs (Yago et al. 1987, Izumida et al. 1988). GETV is widely distributed in Asia, northern Australia, and Far East Russia, where it is maintained by transmission cycles between mosquitoes and domestic pigs (Mair and Timoney 2009). In previous studies, GETV was isolated from various species of mosquitoes, including the genera Culex, Aedes, Armigeres, Anopheles, and Mansonia (Simpson et al. 1975, Igarashi et al. 1982, Bryant et al. 2005). Among them, Cx. tritaeniorhynchus and Ae. vexans are the principal vectors of GETV in Japan and Korea (Kumanomido et al. 1986a, 1986b, Turell et al. 2003). Cx. tritaeniorhynchus is known as the most important vector of Japanese encephalitis virus (JEV; genus Flavivirus, family Flaviviridae) in East Asia, implying that GETV and JEV may cocirculate in a similar transmission cycle in the same habitat. In fact, these two phylogenetically unrelated viruses were isolated together from a single porcine serum sample collected in Kochi, Japan (Tajima et al. 2014).
Historically, GETV outbreaks were reported among racehorses in Japan in 1978, 1979, and 1983 (Kamada et al. 1980, Sugiura et al. 1981, Sentsui and Kono 1985). More recently, in 2014, a sudden GETV outbreak occurred among racehorses in Ibaraki prefecture for the first time in about 30 years (Nemoto et al. 2015, Bannai et al. 2015), and a sequential outbreak at the same site also occurred in the following year (Bannai et al. 2016). Before the outbreak in 2014, GETV appears to have been maintained locally in Japan as antibodies against GETV have been detected sporadically in racehorses at several sites in Japan in 1990s (Sugiura and Shimada 1999). However, the reason for the 2014 outbreak in Japan, which occurred after ∼30 years, is unclear.
During mosquito surveillance studies for JEV in Japan, we unexpectedly obtained multiple GETV isolates from Cx. tritaeniorhynchus mosquitoes collected in Nagasaki in 2012. We determined the whole genome sequence of a GETV isolate, 12IH26, and compared it with those of the previously reported GETV strains. In this study, we discuss the origin and spread of the recent Japanese GETV strains.
Materials and Methods
Mosquito collection and virus isolation
Cx. tritaeniorhynchus mosquitoes were collected in Isahaya city (N32°49′, E130°03′), Nagasaki, Japan, on September 3 and 4, 2012. Blood-fed mosquitoes were collected by an aspirator from 7 PM to 9 PM in pigpens and cowsheds. After digestion of the blood, the Cx. tritaeniorhynchus mosquitoes were sorted by morphological keys. Virus isolation from mosquitoes was performed as previously described (Hoshino et al. 2009).
Viral genome sequencing
To determine the viral genome sequence, we used following next-generation sequencing (NGS)-based techniques. A virus stock (pool No. ID 12IH26) was centrifuged at 1500 rpm for 15 min, and the supernatant was centrifuged again at 8000 rpm for 30 min to remove cellular debris. The fluid was concentrated using a Centriprep Centrifugal Filter Unit YM-50 (Merck Millipore, Billerica, MA) and replaced with SM buffer (100 mM NaCl, 8 mM MgSO4, and 50 mM Tris·HCl). Four units of TURBO DNase (Life Technologies), 3 U of Baseline ZERO DNase (Epicentre, Madison, WI), 25 U of Benzonase nuclease (Merck Millipore), and 0.4 μg of RNase A (Wako Pure Chemical Industries, Osaka, Japan) were added to 390 μL of the concentrate. After incubation at 37°C for 1 h, total RNA was extracted using ISOGEN II (Nippon gene, Tokyo, Japan) and dissolved in 20 μL RNase-free water. The syntheses of the first and second strand cDNAs were performed using cDNA Synthesis Kit (TaKaRa, Shiga, Japan) and cDNA PCR Library Kit (TaKaRa). The PCR-amplified cDNAs were analyzed using the MiSeq system (Illumina, San Diego, CA). CLC genomics workbench software (CLC bio, Aarhus, Denmark) was used to trim adaptors from the sequencing reads and perform de novo assembly for forming the contigs. The contigs were analyzed using BLASTx searches.
The terminal sequences of the viral genome were determined using the rapid amplification of cDNA ends (RACE) techinque. The 5′ RACE was performed using the GeneRacer Kit (Life Technologies) with the gene-specific primer GETV-5RACE-1 (Table 1) for first-strand cDNA synthesis. The 3′ RACE was conducted using the RNA PCR Kit (AMV) Ver.3 (TaKaRa) with the gene-specific primer GETV-3RACE-1 (Table 1). The 5′ and 3′ RACE amplicons were subjected to agarose gel electrophoresis and extracted from the resulting gels. The resultant DNAs were sequenced using an ABI PRISM 3130 Genetic Analyzer (Applied Biosystems) with the specific primers GETV-5RACE-2 and GETV-3RACE-2 (Table 1).
Primer positions are based on the complete genome sequence of the GETV strain 12IH26.
Detection of GETV from the cell culture supernatants
RNA extraction from the virus stocks was performed using the QIAamp viral RNA Mini Kit (QIAGEN, Valencia, CA). Reverse transcription (RT)-PCR for GETV detection was performed using the PrimeScript One Step RT-PCR Kit Ver. 2 (TaKaRa) with a specific primer set for the E2 gene (GETV-E2-FW and GETV-E2-RV; Table 1). The thermal profile consisted of RT at 50°C for 30 min; termination of RT at 94°C for 2 min; and 35 cycles of PCR at 94°C for 30 s, 53°C for 30 s, and 72°C for 2 min. The amplified products were sequenced using the primers used in the RT-PCR and GETV-E2-seq1 (Table 1) as described above.
Analysis of viral genome sequence
The genome sequence of the 12IH26 strain was compared with those of the previously reported GETV strains (Table 2) using GENETYX ver.13 software (GENETYX Co., Tokyo, Japan).
The strain marked by bold is isolated in this study.
Phylogenetic analysis
Molecular phylogenetic analyses were performed based on the nucleotide sequences of the whole genome, E2 gene, partial C gene, and partial nsP1 gene. Multiple alignments of selected viral sequences from GETV strains (Table 2) were conducted using the ClustalW program. Phylogenetic analyses were performed with a maximum-likelihood method using MEGA ver. 7 (Kumar et al. 2015).
Results
Isolation and identification of viruses from Culex tritaeniorhynchus mosquitoes collected in Nagasaki in 2012
A total of 900 female Cx. tritaeniorhynchus mosquitoes were sorted into 42 pooled samples and subjected to virus isolation. Twelve out of 42 samples induced similar cytopathic effects (CPE) with cell shrinkage and aggregation after two blind passages, but only one of them was positive for JEV by RT-PCR assay (data not shown). Therefore we used NGS techniques to identify the infectious agents other than JEV. Pool No. ID 12IH26 was selected as a representative of the CPE-positive samples and analyzed using the Miseq system. A total of 1,145,599 reads were obtained from a cDNA library constructed from the isolate 12IH26. The longest contig resulting from de novo assembly was 11,466 nt in length. It consisted 882,961 reads and its average coverage was 3670.86. This longest contig was highly homologous to the genomic sequence of GETV. The terminal sequences of the viral genome were determined using 5′ and 3′ RACE procedures, allowing the determination of the complete genome sequence. Consequently, the infectious agent present in isolate 12IH26 was confirmed to be GETV and designated GETV strain 12IH26.
Analysis of GETV genome sequence and phylogenetic characterization
The complete genome of GETV strain 12IH26 was 11,689 nt in length. The viral nonstructural (nsP1–4) and structural proteins (C, E3, E2, 6K, and E1) were encoded at positions 79–7482 and 7527–11,288 on the genome, respectively. Like previously described GETV strains, a leaky stop codon was observed upstream of the cleavage site for nsP3/nsP4 in the genome of the 12IH26 strain (data not shown; Shirako and Yamaguchi 2000). A whole-genome sequence comparison of the 12IH26 strain with previously described strains exhibited 97.3–99.9% nucleotide sequence identity (Table 3). The 12IH26 strain was most similar in nucleotide sequence to recent epidemic strains in Japan, 14-I-605-C1 and 14-I-605-C2, which were isolated from horses in Ibaraki, Japan in 2014 (Nemoto et al. 2016). In addition, the amino acid (aa) sequence identities ranged from 99.0% to 99.9%, and the 12IH26 strain had the highest similarity in aa sequence to the strains 14-I-605-C1 and 14-I-605-C2 (Table 3). Amino acid substitutions among the 12IH26, 14-I-605-C1, 14-I-605-C2, and previous GETV strains (MM2021, Sagiyama, M1, MI-110-C1, MI-110-C2, LEIV 16275 Mag, LEIV 17741 MPR, HB0234, South Korea, YN0540, Kochi/01/2005, SC1210; Table 2) were observed in the nsP1, nsP2, nsP3, nsP4, and 6K proteins (Table 4). Almost all of these substitutions in the genome of the 12IH26 strain were identical to those of the strains 14-I-605-C1 and 14-I-605-C2, and most of the substituted aa were concentrated in nsP3 protein. Previous studies have shown that the genomes of several GETV strains contain insertions or deletions in protein-coding or noncoding regions (Wen et al. 2007, Zhai et al. 2008). However, we found no insertions or deletions in the genome sequence of strain 12IH26.
Percentage of nucleotide sequence identities to strain 12IH26.
Percentage of amino acid sequence identities to strain 12IH26.
Positions are based on the amino acid sequence of the GETV strain 12IH26.
GETV strains MM2021, Sagiyama, M1, MI-110-C1, MI-110-C2, LEIV 16275 Mag, LEIV 17741 MPR, HB0234, South Korea, YN0540, Kochi/01/2005, SC1210 (Table 2).
To clarify the phylogenetic position of the strain 12IH26, we performed phylogenetic analyses based on the whole genome, E2 gene, partial C gene (543 nt), and partial nsP1 gene (351 nt) sequences among GETV strains (Fig. 1). In all cases, the 12IH26 strain formed a robust clade with the strains 14-I-605-C1 and 14-I-605-C2 (Fig. 1). Based on the nucleotide sequences of their E2 genes, the strains 12IH26, 14-I-605-C1, and 14-I-605-C2 were closely related to the Chinese strains SH05-15 and SH05-16 (Fig. 1b). Interestingly, phylogenetic analysis using partial C and nsP1 gene sequences showed that the recent Japanese strains 12IH26, 14-I-605-C1, and 14-I-605-C2 formed a clade with the Chinese and South Korean strains, but not with the previous Japanese strains (Fig. 1c, d).
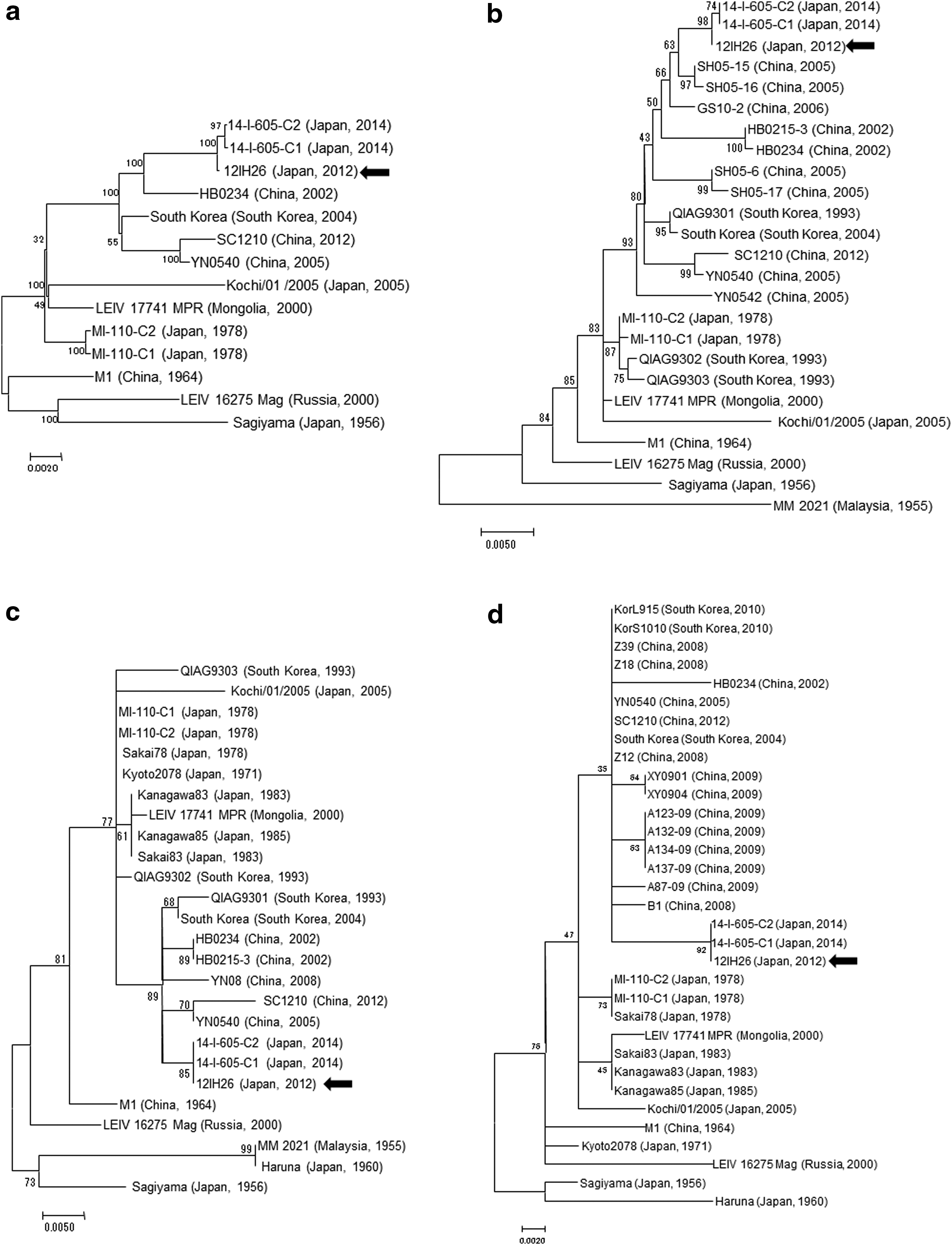
Phylogenetic analyses of GETV strains. Dendrogram of the phylogenetic relationships among the GETV strain 12IH26, other GETV strains, and other related alphaviruses based on their whole genome sequences
GETV prevalence in the Culex tritaeniorhynchus mosquito population in Nagasaki in 2012
The infectious agents present in the 11 CPE-positive samples other than 12IH26 were identified as GETV by RT-PCR assay. Therefore, the minimum GETV infection rate [MIR = (number of positive pools/total specimens tested) × 1000] in Cx. tritaeniorhynchus mosquitoes was calculated to be 13.3. The nucleotide sequences of whole E2 regions of these 11 strains were 100% identical to that of the 12IH26 strain (data not shown). These results suggest that the mosquitoes may have fed on viremic animals (presumably domestic pigs) infected with a genetically homogenous GETV strain circulating in or near our collection site.
Discussion
In this study, we reported the genetic characterization and phylogenetic analysis of GETV strain 12IH26 isolated from Cx. tritaeniorhynchus mosquitoes collected in Nagasaki, Japan in 2012. The collected mosquitoes exhibited a high prevalence of GETV (MIR = 13.3) indicating a high-infection risk in domestic animals, whose value was much higher than those of previous studies in Japan: MIR = 0.28–0.36 in Cx. tritaeniorhynchus mosquitoes and 0.85–2.22 in Ae. vexans nipponii mosquitoes in Ibaraki in 1979 (Kumanomido et al. 1986a), MIR = 0.31–0.72 and 0.87 in Cx. tritaeniorhynchus mosquitoes in Miyazaki and Shiga, respectively, in 1980 (Kumanomido et al. 1986b). However, to date, there are no reports of suspicious or positive cases of GETV infections in pigs, horses, or other animals around our study site in Nagasaki.
Interestingly, the phylogenetic analyses revealed that the strain 12IH26 was most closely related to strains 14-I-605-C1 and 14-I-605-C2, the epidemic strains from the 2014 outbreak at Miho village in Ibaraki, which is about 1000 km away from Nagasaki. In Japan, natural GETV infection in animals is generally thought to occur constantly nationwide, although there are only a few reports on recent GETV prevalence (Sugiura and Shimada 1999, Kyoto biken information for swine veterinarians; available at
Sequence analysis revealed that most of aa substitutions in the 12IH26 strain were observed in the carboxyl-terminal domain of the nsP3 protein and were identical to those of the strains 14-I-605-C1 and 14-I-605-C2. Nemoto et al. (2016) suggested that the mutations in the nsP3 region of the strains 14-I-605-C1 and 14-I-605-C2 might result in altered interactions between GETV and its vectors because carboxyl-terminal domain of nsP3 has been reported to be involved in replication and vector specificity of the genus Alphavirus. In this regard, a recent study revealed that the nsP3 protein of Chikungunya virus (CHIKV; genus Alphavirus, family Togaviridae) interacts with a protein of the host mosquito, which is implicated in the infection efficiency of the virus to the host mosquito (Fros et al. 2015). Therefore, like CHIKV, aa substitutions in the nsP3 of the recent Japansese GETV strains might alter their virological properties. Consequently, it might facilitate the spread of the virus to the mosquito populations, resulting in a high MIR observed in this study. Further studies are needed to characterize the biological properties of these recent Japansese GETV strains in the host mosquitoes.
Phylogenetic analyses showed that the strain 12IH26, 14-I-605-C1, and 14-I-605-C2 are closely related to recent Chinese and South Korean strains, but distantly related to Japanese strains of the 1970s and 1980s. Interestingly, the GETV strain Kochi/01/2005 (derived from a swine serum sample collected in Kochi, Japan in 2005; Tajima et al. 2014) was more closely related to the Mongolian strain than the Chinese strains, South Korean strains, or other Japanese strains. These observations raise the possibility that the recent GETV strains were brought into Japan from overseas at least two times in the past few decades. This may be due to the long-distance migratory property of the vector mosquito Cx. tritaeniorhynchus (Asahina and Noguchi 1968, Ming et al. 1993, Tsuda and Kim 2008). In this regard, molecular epidemiologic studies of JEV have suggested that some JEV strains are imported within Cx. tritaeniorhynchus mosquitoes coming from mainland China to the Japanese archipelago on the west wind (Nabeshima et al. 2009, Sawabe 2014). Therefore, as is the case for JEV, Cx. tritaeniorhynchus mosquitoes infected with GETV might come flying to Japan from mainland China.
Some migratory birds go back and forth between Japan and the Korean Peninsula, northeastern China, Mongolia, or Far East Russia. Sometimes they transport avian influenza viruses (genus Influenza virus A, family Orthomyxoviridae) to Japan (Sakoda et al. 2012). As mentioned above, phylogenetic analyses showed that the GETV strain Kochi/01/2005 is closely related to the Mongolian strain, suggesting that this strain might be transported into Japan by migratory birds. Scherer et al. (1962) reported that black-crowned night herons (Nycticorax nycticorax, a migratory bird species) in heronries near Tokyo had neutralizing antibodies to SAGV, a strain of GETV originally isolated from Cx. tritaeniorhynchus mosquitoes in the same sites. In addition, in another study, antibodies against GETV were detected in sera from birds (Doherty et al. 1966), suggesting potential vertebrate hosts of GETV. Thus, GETV might also be brought into Japan by infected migratory birds; however, no studies to date have assessed the GETV infection status of the migratory birds. Future seroepidemiologic study of wild birds might help to understand the long-distance spread and persistence of GETV.
Other than migrating birds and mosquitoes, mosquitoes aboard aircraft have been identified as the greatest threat for West Nile virus (genus Flavivirus, family Flaviviridae) introduction to an island by the quantitative risk assessment of various pathways, such as human-transported mosquitoes, wind-transported mosquitoes, human-transported birds, and migratory birds (Kilpatrick et al. 2004, 2006, Douglas et al. 2007, Brown et al. 2012). Although the same risk-assessment framework is applicable to GETV introduction, data on the transport of goods and animals and especially the epidemiology of GETV are insufficient currently, and an intensive seroepidemiologic study of wild birds will be required in future.
Conclusions
Our genetic and phylogenetic analyses of GETV strains revealed that the strain 12IH26 from Nagasaki in 2012 is closely related to the epidemic strain from the 2014 outbreak in Ibaraki and may have originated from a recent GETV strain in mainland China or the Korean Peninsula. Furthermore, it was also suggested that GETV may be transported into Japan through long-distance migrations of mosquitoes or migratory birds from overseas. More information about the complete genome sequences of GETV strains from both mosquito vectors and reservoir hosts will be important for understanding the processes involved in the introduction and spread of GETV in Japan.
Footnotes
Acknowledgments
This work was supported by grants from the Japanese Ministry of Health, Labor and Welfare (24-Shinko-Ippan-007) and the Research Program on Emerging and Re-emerging Infectious Diseases from Japan Agency for Medical Research and Development, AMED.
Author Disclosure Statement
No competing financial interests exist.