
Abstract
Hantaviruses are RNA viruses primarily carried by rodents, soricomorphs, and bats. The data about the distribution and genetic diversity of these viruses are often limited, especially in most regions of sub-Saharan Africa. Moreover, the majority of representatives were identified in western African localities, while only three hantaviruses have been reported in East Africa to date. In this study, a total of 1866 small mammals captured between 2009 and 2014 in various countries of Eastern Africa (Ethiopia, Zambia, Mozambique, Kenya, and Tanzania) were molecularly screened for the presence of hantaviruses. Hantavirus RNA was detected in dried blood samples of the Cape pipistrelle bat (Neoromicia capensis) captured in Ethiopia and the African wood mouse (Hylomyscus endorobae) from Kenya. Phylogenetic analysis of partial genomic segments revealed that the Ethiopian sample represents a sister lineage of the Mouyassué virus (reported previously from the congeneric bat in Côte d'Ivoire), and the Kenyan sample is a sister lineage of the Sangassou virus (described from the same mouse genus in Guinea).
Introduction
H
Materials and Methods
Within biodiversity surveys in several regions of Eastern Africa, a total of 1866 small mammals (rodents, shrews, and bats) were captured across various habitats in 2009–2014. In total, 224 small mammals from Zambia, 269 from Mozambique, 528 from Kenya, 345 from Ethiopia, and 500 from Tanzania were trapped and analyzed (Supplementary Table S1; Supplementary Data are available online at
Results
Hantaviral RNA was detected in dried blood samples of two animals: one Cape pipistrelle bat (Neoromicia capensis) from a total of nine individuals of this species from Dhati Walel National Park in Ethiopia (N 09°13′33″ E 34°52′37″, elevation 1427 m, captured in February 2014), and one African wood mouse (Hylomyscus endorobae) from a total of five individuals of this species from Mount Kenya (S 0°9′40.8″ E 37°26′45.72″, 3000 m, July 2010). After Sanger sequencing, sequences were manually shortened due to their reduced quality at both ends: we thus obtained a 237 nucleotide-long sequence from the Ethiopian hantavirus and a 335 nucleotide-long sequence from the Kenyan hantavirus. The sequences were first BLASTed to check if they belonged to hantaviruses. The fragments were then aligned and compared with selected hantavirus sequences available in GenBank. Preliminary phylogenetic analysis suggested that the Ethiopian virus is related to Mouyassué virus (MOUV) (Sumibcay et al. 2012), and the sequence from Kenya was close to Sangassou virus (SANGV) (Klempa et al. 2006). To obtain additional parts of both hantavirus genomes, PCR primers designed for MOUV (Gu et al. 2014) and SANGV (Klempa et al. 2012) were used. Repeated attempts to amplify additional parts of the Ethiopian virus genome were unsuccessful, while partial sequences of the M and S segments (288 bp and 529 bp, respectively) were obtained for the hantavirus from Kenya (GenBank KX184827-KX184830).
The phylogenetic analysis of partial L segment sequences confirmed that the hantavirus from Ethiopian Neoromicia capensis is sister to MOUV described from Neoromicia nanus in Côte d'Ivoire (posterior probability, PP = 0.99) (Fig. 1). Kenyan hantavirus from Hylomyscus endorobae grouped with the well-known Murinae-associated hantaviruses and forms a sister lineage to two strains of SANGV detected from Hylomyscus simus in Guinea (PP = 0.75). The sister relationship with SANGV was validated by analysis of partial M segment sequences (PP = 1), while for the S segment the partial sequence formed a sister lineage to well-supported cluster of Dobrava–Belgrade virus, Saaremaa virus, and SANGV (PP = 1). Genetic distances among nucleotide and amino acid sequences of obtained strains compared to closely related hantaviruses are given in Supplementary Table S2. The distance values showed significant differences between the new found viruses and their closest relatives, suggesting that in fact they may even represent new hantavirus species.
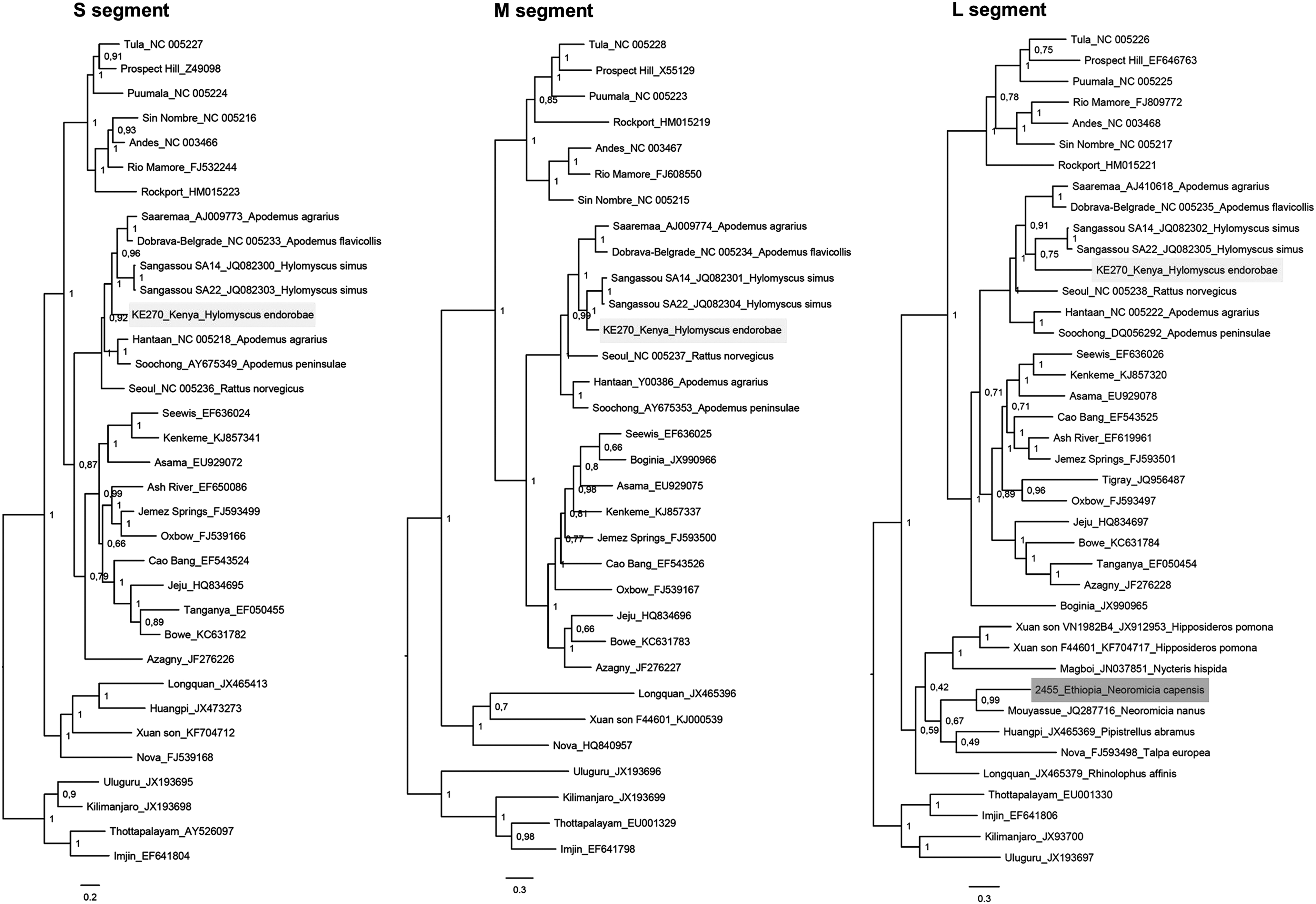
Bayesian phylogenetic trees of newly sequenced strains from Eastern Africa and representatives of hantaviruses for the S, M, and L segments (GenBank accession numbers are provided after the virus names). Analyses were performed in MrBayes on XSEDE in CIPRES Science Gateway Webserver; numbers above nodes indicate the Bayesian posterior probability values. Partial hantavirus sequences from the Kenyan Hylomyscus endorobae are highlighted by light gray background and partial sequence of L segment from the Ethiopian Neoromicia capensis is marked with dark gray background.
Discussion
Although hantavirus discoveries were for a while mainly restricted to Western Africa (review in Witkowski et al. 2014), the present investigation indicates that certain West African hantaviruses may have counterparts circulating in East Africa and that the lack of hantavirus records outside Western Africa is rather related to insufficient research intensity. The presence of sister hantavirus lineages in the Hylomyscus rodents in both Western and Eastern Africa may be related to the phylogeography and phylogeny of these small mammals. Genus Hylomyscus is distributed in tropical forests with many species recorded from eastern African mountain forests (with H. endorobae belonging to the basal group of the genus), through the Congo basin to Guinean forests in westernmost Africa (from where H. simus was described) (Nicolas et al. 2006; Nicolas et al., pers. comm.). It seems, therefore, highly probable that future search will lead to discoveries of SANGV-related hantaviruses in other parts of tropical Africa.
The distribution range of Neoromicia capensis spreads over sub-Saharan Africa from Guinea to Kenya and most of South Africa, overlapping in many parts with the range of Neoromicia nanus (Happold and Happold 2013). In view of this geographical context, we can suppose these small insectivorous bat species could disseminate MOUV over long distances.
Conclusions
The present report describes the occurrence of sister lineages of two known western African hantaviruses in Eastern Africa, which evidence that their geographic range is much larger than previously expected. Even though presence of viruses similar to western African hantaviruses was corroborated in Ethiopia and Kenya, only small parts of genomes were obtained limiting relevant phylogenetic analysis. Further efforts to gain the full genomes of SANGV- and MOUV-related viruses, detailed phylogenetic analysis and thorough investigation of hantavirus cophylogeography in conjunction with their hosts are now required to correctly understand the evolution of African hantaviruses and to predict potential threats to human health.
Footnotes
Acknowledgments
This work was supported by the Czech Science Foundation (GACR grant P502/11/J070) and the Russian Foundation for Basic Research (project no. 15-04-03801-a). For permission to collect specimens, we are beholden to the National Research Council and Forestry Department in Malawi, the National Council for Science and Technology, the Ethiopian Wildlife Conservation Authority, the Oromia Forest and Wildlife Enterprise (Ethiopia), the Zambian Wildlife Authority, the Kenyan Forest Service and the Kenyan Wildlife Service, the National Directorate for Protected Areas (DINAC—Mozambique), and the Sokoine University of Agriculture in Morogoro (Tanzania). We acknowledge also numerous collaborators who helped to collect small mammals in particular countries, especially Radim Šumbera, Sophie Gryseels, Vladimír Mazoch, and Hana Patzenhauerová-Konvičková. We sincerely thank Dr. Richard Yanagihara for providing the primer sequences to obtain additional parts of Mouyassué relative genome, and Eva Rybníčková for the help with screening of Ethiopian samples.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.