
Abstract
Aedes-borne viruses are responsible for high-impact neglected tropical diseases and unpredictable outbreaks such as the ongoing Zika epidemics. Aedes mosquitoes spread different arboviruses such as Dengue virus (DENV), Chikungunya virus (CHIKV), and Zika virus, among others, and are responsible for the continuous emergence and reemergence of these pathogens. These viruses have complex transmission cycles that include two hosts, namely the Aedes mosquito as a vector and susceptible vertebrate hosts. Human infection with arboviruses causes diseases that range from subclinical or mild to febrile diseases, encephalitis, and hemorrhagic fever. Infected mosquitoes do not show detectable signs of disease, even though the virus maintains a lifelong persistent infection. The infection of the Aedes mosquito by viruses involves a molecular crosstalk between cell and viral proteins. An understanding of how mosquito vectors and viruses interact is of fundamental interest, and it also offers novel perspectives for disease control. In recent years, mass spectrometry (MS)-based strategies in combination with bioinformatics have been successfully applied to identify and quantify global changes in cellular proteins, lipids, peptides, and metabolites in response to viral infection. Although the information about proteomics in the Aedes mosquito is limited, the information that has been reported can set up the basis for future studies. This review reflects how MS-based approaches have extended our understanding of Aedes mosquito biology and the development of DENV and CHIKV infection in the vector. Finally, this review discusses future challenges in the field.
Introduction
A
To date, no specific antiviral drugs and no effective vaccine platform for arboviruses exists. A vaccine against YFV is uniquely successful, and it has been applied to humans for decades. Recently, the tetravalent DENV vaccine (CYD-TDV) was licensed in several countries; however, it renders only partial protection against the infection and is associated with an unexplained increased incidence of hospitalization for severe dengue disease among children who are younger than 9 years (Hadinegoro et al. 2015). In consequence, vector control remains the primary approach to prevent dengue and other widespread arbovirus infections of humans worldwide.
Historically, Aedes aegypti and Aedes albopictus are the two species responsible for most diseases transmitted. Originating in Africa, Ae. aegypti and in Asia, Ae. albopictus are now present globally and exhibit a tropical and subtropical distribution. Most notably, their habitats are closely associated with humans, which has made them an unusually successful vector. Female mosquitoes gain the viruses from infected person's blood and after an incubation time, they spread the virus while feeding on another person. Aedes mosquitoes that are infected by viruses do not show detectable signs of illness, even though the mosquito remains infective for the rest of its life. In some regions in Asia and Africa, the transmission cycle may also involve jungle primates that act as a reservoir for the virus (Guzman and Harris 2015, Higgs and Vanlandingham 2015). Many factors facilitate the spread of vectors-viruses to new geographic regions, but the most important are urbanization, globalization, and a lack of effective mosquito control (Gubler 2011).
The intrinsic ability of an arthropod vector to acquire, maintain, and transmit a pathogen is defined as vector competence (VC) (Bennett et al. 2002, Carrington et al. 2013). Interactions between mosquitoes and viruses are vector-specific and virus strain-specific, although Aedes spp. have different levels of VC to transmit viruses. Some of this specificity derives from the ability of a particular arbovirus to overcome tissue barriers in the vector to establish a persistent infection. In addition, infection of the Aedes mosquito by viruses involves molecular crosstalk between insect cells and viral proteins as the result of two different forces: pro-virus replication and the antiviral response. Strategies of mosquito control require comprehensive knowledge of the molecular interactions between a virus and a vector and an understanding of the complex factors that determine VC.
The continuous advances in all types of techniques for genome-wide analyses have driven the study of vector-borne diseases into a new era, the big data science posgenomic. The characterization of proteins, peptides, and lipids in the Aedes vector contributes to the understanding of the biology of mosquitoes, knowledge of the processes that controls virus replication in mosquito tissues, and the generation of plausible new strategies to prevent and control virus transmission. Several comprehensive mass spectrometry (MS)-based studies have been performed to identify the components of mosquito cells or tissues, and significant effort has been expended toward the discovery of proteins, signal pathways, and protein–protein interaction networks that are present during mosquito virus infection. This review focuses on MS-based strategies, contributions, challenges, and future perspectives and its potential to add valuable information to the understanding of Ae. aegypti biology and its interactions with two important human viral pathogens, DENV and CHIKV.
Mosquito Vector
The Aedes genus includes more than 900 species of mosquitoes worldwide (Gaffigan et al.). Although they are important disease vectors, the diversity and geographic distribution of mosquito species in the different zones of the world are poorly known. Of these species, the main vector of DENV is the mosquito Ae. aegypti, which lives in tropical and subtropical areas in close association with human habitats. The secondary vector for DENV is the Asian tiger mosquito Ae. albopictus, which is considered the most invasive Aedes spp. Ae. albopictus colonizes diverse habitats, even cold temperature regions, and replaces local species (Paupy et al. 2009).
Given the importance of these mosquitoes in disease transmission, Ae. aegypti is a model organism with which to study mosquito biology that allows the investigation of multiple mosquito–pathogen interactions. This species has 15,419 genes in a genome size of 1.38 Gbp, of which nearly half (47%) is composed of transposable elements (Nene et al. 2007), and there are several nuclear copies of the mitochondrial origin genome (Black and Bernhardt 2009, Hlaing et al. 2009). Studies of population genetics revealed a varying degree of worldwide genetic polymorphism (Urdaneta-Marquez and Failloux 2011, Manni et al. 2015). Recently, a single nucleotide polymorphism (SNP) chip was developed to screen 50,000 SNPs present in Ae. aegypti populations around the world (Evans et al. 2015). The tremendous genetic variability of Ae. aegypti is a challenge in understanding the evolutionary history, competence as a disease vector, and for the study of the effects and efficacy of vector control methods. On the other hand, Ae. albopictus has the largest mosquito genome, and the recent report of its genome sequence (Chen et al. 2015), shows that it consists of 1.967 Gbp with 68% transposable elements and ∼40% more DNA than Ae. aegypti. The availability of genomic sequence data for Ae. aegypti and Ae. albopictus mosquitoes provides the basis for proteomics profiling analysis, which reveals information on protein expression in response to a variety of stimuli including virus infection.
Virus Replication Cycle in the Mosquito
Aedes-borne viruses infect vertebrate and invertebrate hosts, and for this alternation, viruses have developed the ability to modulate diverse intracellular environments and to overcome different types of antiviral responses. The persistent arbovirus infection of an Aedes mosquito requires the successful crossing of tissue barriers by the virions and modulation of the innate immunity viral response (Hardy et al. 1983, Franz et al. 2015). First, the mosquito ingests viremic blood, and then viruses initiate the infection in the midgut, an immunocompetent organ that represents the first and most challenging barrier for VC (Black et al. 2002). After sufficient viral replication is carried out in the midgut epithelium, viruses pass across the basal lamina of the gut epithelium, and virions spread through the hemolymph to other tissues such as the fat body, trachea, and others (second barrier), where secondary amplification occurs; then, the virus infects the salivary glands (SGs; third barrier). Once replication has proceeded to the SGs, the virus is secreted into the mosquito salivary ducts (fourth barrier) for horizontal transmission to an uninfected vertebrate host during a subsequent blood-feeding event (Black et al. 2002, Bennett et al. 2005). This process is known as the extrinsic incubation period (EIP), and it can vary depending on conditions such as mosquito strain, virus strain, and temperature, but it ranges from 7 to 14 days (Arias-Goeta et al. 2013, Tjaden et al. 2013). The infectious process in mosquitoes has been characterized principally by DENV. Interestingly, the CHIKV EIP is short, ranging from 2 to 6 days, and produces high viral titers in the mosquito (Dubrulle et al. 2009). The mechanism that arboviruses employ to disseminate from the midgut to other tissues remains elusive. It has been hypothesized that these viruses may induce the activity of proteases and apoptosis to overcome tissue barriers in the mosquito (O'Neill et al. 2015).
On a molecular level, although DENV and CHIKV replicate in Aedes mosquitoes, these viruses have different replication strategies. DENVs are members of the Flavivirus genus and Flaviviridae family and have a genome that is composed of positive single-stranded RNA (+RNA) that encodes a single open reading frame (ORF), which is flanked by highly structured 5′ and 3′ untranslated regions (UTRs). Interestingly, the flavivirus adaptation process appears to depend on the selection of 3′UTR RNA that leads to a fitness increase in the host (Villordo et al. 2015). The DENV genome is translated as a single polyprotein and is subsequently cleaved into three structural proteins (capsid-C, envelope-E, and prM) and seven nonstructural proteins (NS1, NS2A/B, NS3, NS4A/B, and NS5). Structural proteins determine viral serotypes and are involved in viral attachment and entry. Nonstructural proteins are essential for viral replication, and they are largely conserved across DENV serotypes (Perera and Khun 2008). The CHIKV is a member of the Togaviridae family and genus Alphavirus. The CHIKV genome is composed of positive single-stranded RNA with two ORFs. The first ORF is translated directly from genomic RNA that encodes four nonstructural proteins (NSP1 to NSP4) that are required for RNA synthesis. The second ORF is translated from a subgenomic 26S RNA as a polyprotein that encodes five structural proteins (capsid-C, envelope-E1, envelope-E2, two proteins with ion channel activity 6K and E3). The structural proteins have functions in the assembly of new viral particles and during the attachment and entry into cells (Jose et al. 2009).
The viral replication cycle of DENV and CHIKV has primarily been studied in mammalian cells. The viral cycle starts with viral particle attachment to one of the ubiquitously expressed attachment factors or receptors on the cell surface. These viruses have a widespread tropism in the invertebrate and vertebrate host that has resulted in some candidates of cellular molecules being proposed as receptors (Hidari and Suzuki 2011, van Duijl-Richter et al. 2015). Nevertheless, the entry receptor for DENV or CHIKV infection in the mosquito Aedes has not been identified. Subsequently, the virus is internalized into the cell via clathrin-mediated endocytosis (Hase et al. 1989, Lee et al. 2013), and the RNA genome gains entry into the cytoplasm via viral glycoprotein-mediated membrane fusion. The RNA genome is translated by host cell machinery, and structural and nonstructural viral proteins guarantee that replication will occur in different cellular types so that the transmission cycle proceeds.
DENV replication occurs on modified endoplasmic reticulum (ER) membranes, and it depends on the infected cell lipid metabolism (Welsch et al. 2009). Following RNA replication and synthesis of the viral structural proteins, immature virions are assembled via the budding of newly formed nucleocapsids inside the lumen of the ER. During this process, the virus particles obtain their lipid bilayer envelope and the particles mature by passing through the Golgi and trans-Golgi network. Finally, progeny virus particles are released from the cell via exocytosis (Perera and Khun 2008). Multiple efforts have been realized to identify the host factors that are required for DENV replication in human and insect cells. While in humans it is known that DENV infection induces apoptosis, the unfolded protein response (UPR), autophagy and activation of the ubiquitin proteasome system (Fischl and Bartenschlager 2011, Acosta et al. 2014), in the invertebrate host, the modulation of these processes during DENV infection has not been reported. Recently, was reported that a putative 3′-5′ RNA exonuclease (ERI3) associates with DENV genomic RNA in human and insect cells; this protein is required for the accumulation of viral RNA and the production of infectious particles in both hosts (Ward et al. 2016).
On the other hand, the analysis of CHIKV replication in humans has greatly advanced after recent global outbreaks, but the information about the replication cycle in the mosquito is limited. It is known that alphavirus replication induces rearrangement of host membranes into cytoplasmic structures known as cytopathic vacuoles (CPVs) (Frolova et al. 2010, Spuul et al. 2011). RNA synthesis occurs on the cytoplasmic side of CPVs, although the mechanism of formation and movement of higher order membrane structures remains unclear. However, the cytoskeletal association of viral nonstructural protein 3 (nsP3) suggests that it plays a role in the formation of CPVs (Frolova et al. 2006, Gorchakov et al. 2008). Recently, it has been demonstrated that the interactions of CHIKV nsP3 with the vertebrate host Ras-GAP SH3 domain-binding protein (G3BP) prevent the assembly of stress granules during infection and modulate the mammalian stress response (Fros et al. 2012). Interestingly, in mosquitoes, CHIKV nsP3 localized in the cytoplasmic granules in association with Rasputin (Rin), a G3BP homolog protein in Aedes mosquitoes. Rin depletion in vivo decreased the CHIKV infection rate and the transmissibility in Ae. albopictus, which suggests a role of Rin as an important proviral determinant for CHIKV infectivity (Fros et al. 2015).
Arbovirus replication cycle in the invertebrate host has been few explored at the molecular level. These viruses produce undetectable pathology in the infected mosquitoes, which tolerate persistent infections and exhibit lifelong virus transmission (Blair 2011). In nature, Aedes mosquitoes are exposed to different pathogens, and upon infection, they mount a strong innate immune response that differs substantially from the human response; this dissimilarity could be the basis for the contrasting consequences of infection in the two hosts. Due to the intrinsic properties of a particular virus, and considering the major evidence about the replication cycle in mammalian cells, it is logical to think that the requirements for viral replication can be different for each virus in the mosquito vector.
MS Strategies
MS is an analytical technique used to measure the molecular mass of a chemical compound. The complete process involves the conversion of the sample into gaseous ions, with or without fragmentation, which are then characterized by their mass to charge ratios (m/z) and relative abundances. In the last 25 years, the advent of MS coupled with sequencing genome projects and the development of bioinformatics has benefited MS-based strategies, which enable the large-scale identification of proteins, peptides, lipids, and metabolites. Advances in protein multidimensional separations as well as progress in speed, analytical sensitivity, and dynamic range in MS have improved the scale of experiments for proteomics (Sabidó et al. 2012). A new era of quantitative biology based on MS is underway. It is now possible to define the content, relative abundance, and post-translation modifications (PTMs) of proteins and interaction partners of proteins in a dynamic fashion to address biological questions.
Traditionally, investigations in proteomics have used two-dimensional gel electrophoresis (2-DE) for protein separation of heterogeneous protein samples based on isoelectric point, molecular mass, solubility, and relative abundance. However, there are some limitations that are inherent to this technique, such as the incapacity to resolve extremely basic, acidic, and hydrophobic proteins, which leads to a low dynamic range for relative quantification and is time consuming for reproducible results (Rabilloud and Lelong 2011). The use of differential in-gel electrophoresis (DIGE) could resolve some of these disadvantages. In this methodology, 2-DE has been combined with protein labeling strategies using up to three different fluorescent Cy-dyes (Cy2, Cy3, and Cy5) to allow comparative analysis of different protein samples within a single 2-DE gel with a higher sensitivity and at a higher linear range (Marouga et al. 2005). However, methodological advances in the last decade have provided more sensitive and high-throughput nongel-based techniques, including the following: (1) Stable isotope labeling by amino acids in cell culture (SILAC), which depends on the incorporation “in vitro” of specific isotopic-labeled amino acids that can tag cell proteins and peptides. The light and heavy forms of individual peptides are chemically identical; they are simultaneously detected during MS analysis, and their peak intensities are then compared to determine the change in abundance in one sample relative to that of the other sample (Ong et al. 2002, Zhu et al. 2010). Methods to isotopically label proteins or peptides include metabolic labeling of model organisms such as Drosophila (Sury et al. 2010) and the rat (McClatchy et al. 2011). (2) Isobaric Tag for Relative and Absolute Quantification (ITRAQ); in this technique, the N-terminuses of peptides that are obtained from protein digestions are covalently labeled with varying mass tags. The tags are then cleaved from the peptides via collision-induced dissociation during MS/MS, which is required for this type of quantitative proteomic analysis. Peptides with different tags are indistinguishable by mass but can be differentiated in MS/MS spectra through the release of a reporter ion; each represents a different mass (114, 115, 116, or 117 Da). Four or eight samples can be differentially labeled, and the intensity of reporter ions allows for the simultaneous sequencing and quantification of labeled peptides (Thompson et al. 2003, Rauniyar and Yates 2014). The limitations of labeling techniques include the high cost of the reagents, the higher concentration of sample that is required, and incomplete labeling. These restrictions have led to the development of label-free methods for protein quantitation as rapid and low-cost alternatives. (3) Label-free liquid chromatography (LC)/MS is based on the observation that signal intensity from electrospray ionization correlates with ion concentration. Consequently, the relative peptide levels between samples can be determined directly from these peak intensities. Protein quantitation is performed via ion peak intensity or spectral counting in the MS/MS analysis. As a result of the large amount of data acquired from these experiments, sensitive computer algorithms are required for automated ion peak alignment and comparisons (Chelius and Bondarenko 2002, Florens and Washburn 2006). The benefits of these new methodologies have not been fully exploited in Aedes mosquito investigations. Nevertheless, several proteomics studies both in vivo and in vitro have been performed to better understand Aedes mosquito basic biology and its interactions with various pathogens. In the following section, we discuss contributions and challenges that this field is currently facing in terms of the application of proteomics to the analyses of virus–vector interactions and the potential implications for understanding disease transmission.
Proteomics to understand the biology of mosquitoes
Knowledge of mosquito physiology is critical to find ways to prevent pathogen transmission to human populations. Research on this topic has been focused on mosquito components that are involved in survival and/or molecules that are involved in the infection process.
Mosquito eggshells exhibit diverse physical properties and have a structure that is consistent with adaptations to the different environments where these insects live. The survival of Ae. aegypti embryos through the adverse environment is crucial to complete the life cycle and therefore for the persistence of mosquito populations. Studies of eggshell formation and protein composition have been performed with proteomics and transcriptomics approaches (Yao and Li 2003, Marinotti et al. 2014). A total of 130 proteins were identified from the Ae. aegypti eggshell; of these, 3 novel vitelline membrane proteins were discovered, along with odorant-binding and cysteine-rich proteins that may be structural components of the eggshell. Additionally, enzymes with peroxidase, laccase, and phenoloxidase activities were also identified. Therefore, MS was used to identify specific proteins that are involved in chorion metabolism such as melanization proteins and chorionic protein cross-linker (Kim et al. 2005). This valuable knowledge may lead to novel methods with which to interfere in mosquito reproduction and may consequently reduce vector-borne transmission.
Another relevant aspect of mosquito reproduction is the protein composition of seminal fluid, which is transferred from males to females during mating. Seminal soluble proteins (Sfps) are crucially important for male reproductive success, and they modulate several aspects of female postmating behavior and physiology, including egg production and blood feeding (Avila et al. 2012). Because of its importance in the mosquito life cycle, the semen proteome from Ae. aegypti and Ae. albopictus has been characterized. Initially, 250 proteins expressed in the male reproductive glands of Ae. aegypti mosquitoes were described using bioinformatics and proteomics approaches and 53 of those proteins were considered putative Sfps based on the sequences secretion signal prediction (Sirot et al. 2008). To identify male-derived proteins that are transferred to females during mating, the researchers adapted a SILAC technique for the in vivo labeling of Ae. aegypti proteins with the objective to distinguish male- and female-derived proteins in the mated female reproductive tract (Sirot et al. 2011). The principle of this method is to mate males to females whose proteins are labeled with the stable isotopes to exclude the female proteins from MS identification. This approach allowed identifying 145 Sfps, 123 are newly recognized components of Ae. aegypti semen, 84 were previously identified from the reproductive glands, however, they were not designated as putative Sfps because they lacked predicted secretion signal sequences (Sirot et al. 2008). The remaining 22 proteins are transferred to females during mating, biological function of these proteins suggest roles in protein activation/inactivation, ecdysteroidogenesis, and sperm utilization (Sirot et al. 2011). Sfps studies have revealed an asymmetric pattern of reaction to these proteins. Sfps from Ae. albopictus induce typical postmating changes in recipient Ae. aegypti females. In contrast, donor Sfps from Ae. aegypti have little or no effect on recipient Ae. albopictus females, which suggests that these proteins have an effect on postmating behavior, including host seeking, egg development and deposition, and refractoriness to mating (Lee and Klowden 1999, Tripet et al. 2011, Bargielowski et al. 2013). Transcriptomic and proteomic approaches have been used to characterize the seminal fluid of Ae. albopictus (Boes et al. 2014). The authors found 314 proteins that are synthesized in male Ae. albopictus and are moved to females during mating; interestingly, 198 are putative Sfps. The majority of the Ae. albopictus Sfps (134) are derived from transcripts that are found in both the male and female reproductive tracts, and the remaining one-third (64) are derived from transcripts that are found exclusively in males. Interestingly, this discovery highlights the benefit of the use of a proteomics approach to identify Sfps rather than male-specific gene expression (Boes et al. 2014). Sequence comparison of these Sfps with proteins in several other insects, including Ae. aegypti, showed that only 72 (36.4%) Ae. albopictus Sfps have putative orthologs in Ae. aegypti, which suggests a low conservation of the complement of Sfps in these species. Functional analysis of the differential Sfps could guide strategies for the management of the rate of pathogen transmission by Ae. albopictus.
Mosquito longevity is another critical factor that affects mosquito-borne pathogen-transmission cycles and vectorial capacity (Iovinella et al. 2015). Recently, two studies that used quantitative proteomics evaluated the relative abundance of the mosquito proteins during aging. In the first study, the authors used Ae. aegypti obtained from a recently colonized laboratory strain (Hugo et al. 2013) and found 10 proteins that changed in expression level during the life of the mosquito. They reported four proteins, ADF (actin depolymerizing factor); eIF5A (eukaryotic initiation factor 5A); Q17LN8 (insect cuticle protein [ICP]), and AFP (anterior fat body protein) as potential aging markers. In the second study, samples from Ae. albopictus eggs at different development stages from field-collected organisms were evaluated via proteomics methods with label-free LC/MS/MS (Iovinella et al. 2015). The authors identified and quantified 1000 proteins and proposed seven mosquito proteins as possible markers for age-grading for field populations. These proteins are GST (glutathione S-transferase), ICP, PC (pyruvate carboxylase), AAEL001319-PA (a 25 kDa protein), ADF, and SCP-1 AAEL005961-PA.
The natural entry of arboviruses in the mosquitoes is the midgut, which constitutes the most important barrier of virus replication and is the main active immunological tissue to limit subsequent viral dissemination. Given their importance different authors reported the characterization of proteins in the midgut of the female mosquito. Popova-Butler and Dean (2009) isolated brush border membrane vesicle (BBMV) proteins from Ae. aegypti larval midguts and used two proteomic methods: 2D gel electrophoresis and a shotgun two-dimensional liquid chromatographic approach based on multidimensional protein identification technology (MudPIT). These authors identified 119 proteins. Thirty-six proteins were detected by both methods in the Ae. aegypti BBMV. Three proteins, namely arginine kinase, putative allergen, and actin, are the most abundant proteins in the sample. The first study to address the midgut proteome evaluated Ae. albopictus females that were fed only with sugar. Fifty-six proteins were identified by comparison with the Ae. aegypti genome and were classified into 15 categories according to GO annotation at the VectorBaseDB. The most abundant groups of proteins were associated with amino acid metabolism, unknown biological processes, and cell redox homeostasis (Saboia-Vahia et al. 2012). In another investigation, the characterization of peptidases in the midgut of sugar-fed Ae. albopictus females was performed (Saboia-Vahia et al. 2014). The authors reported the expression of active trypsin-like serine peptidases; however, only three peptidases were identified. This descriptive information contributed to the functional annotation of Aedes spp. genomes through the use of MS data.
The mosquito SG represents the final route for virus transmission and is the second tissue in terms of importance during virus replication. Arthropods secrete a large number of bioactive molecules in the SG for efficient blood feeding, and these molecules can modulate host hemostasis (platelet aggregation, blood clotting, and vasoconstriction), inflammation, and the immune response (Ribeiro and Valenzuela 2003). Mosquito saliva is able to enhance viral infections, and saliva components may differ from one species to another and between mosquito genera (Schneider et al. 2006, Calvo et al. 2009, Conway et al. 2014). However, the mechanism and saliva proteins that are involved in enhanced transmission are not known. The SG transcriptomes of male and female mosquitoes were analyzed, and the authors reported that Ae. aegypti saliva contains over 100 unique proteins that have been classified as D7 proteins, phosphatidylethanolamine binding proteins, odorant and juvenile hormone binding proteins, serpins and other protease inhibitors, a sialokinin vasodilator, nucleotidases, serine proteases, lectins, angiopoietins, antimicrobial proteins and peptides, mucins and peritrophins, and some proteins of unknown function (Ribeiro et al. 2007, 2016). However, functional data are not available for the majority of these proteins. Interestingly, a proteomics study was performed to compare the sialome of three Ae. aegypti mosquito colonies (PAEA, Rockefeller, and Formosus) using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) DIGE and MS (Almeras et al. 2010). The authors identified 120 distinct SG proteins. More than 50% of these proteins have a cytoplasmic and mitochondrial localization, and 12.5% (15) are secreted proteins; of these, 11 proteins were involved in mosquito blood feeding or in the regulation of blood coagulation, and the other proteins have unknown functions. Finally, no difference was detected between the three colonies. On the other hand, the saliva of mosquitoes induces antibody production in people who live in regions with higher Ae. aegypti populations (Doucoure et al. 2012). Recently, two bands of immunogenic proteins (56 and 31 kDa) were detected in serum samples from dengue patients and healthy people of the endemic region in comparison with serum samples from individuals who lived in a nonendemic region. Of those bands, 13 proteins were characterized from the 31-kDa immunogenic proteins and 7 proteins were characterized from the 56-kDa immunogenic proteins. Among these, the most abundant are the D7 protein (Arthropoda Odorant-Binding Protein, AOBP) and apyrase (Oktarianti et al. 2015). Other proteomics efforts to elucidate the biology of Aedes vector mosquitoes have included the comparison of proteins and phosphoproteins that were isolated from the Malpighian tubules of females with and without stimulation with a diuretic peptide, the authors reported that insect kinins appear to active paracellular and transcellular transport pathways (Beyenbach et al. 2009). The published subproteome of Aedes mosquito, as described above, is summarized in Table 1.
2-DE, two-dimensional gel electrophoresis; ADF, actin depolymerizing factor; AFP, anterior fat body protein; BBMV, brush border membrane vesicle; DIGE, differential in-gel electrophoresis; GST, glutathione S-transferase; ICP, insect cuticle protein; LC, liquid chromatography; MS, mass spectrometry; MudPIT, multidimensional protein identification technology; PC, pyruvate carboxylase; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis; Sfps, seminal fluid proteins; SILAC, stable isotope labeling by amino acids in cell culture.
Proteomics to study the host infectome
In the following section, we describe some analyses of interactions between the Aedes mosquito and CHIKV or DENV in which proteomics approaches were applied (Table 2).
The identification of host proteins that are critical for pathogen infection via MS-based proteomics in combination with bioinformatics has become a powerful methodology for the characterization of the host infectome. Modern proteomic techniques have recently increased the number of identified host factors involved in virus infection and have identified biological processes and cellular regulatory networks involved in viral replication. DENV or CHIKV infection in mammalian host cells has mainly been studied with a proteomics approach (Chiu et al. 2014, Pando-Robles et al. 2014, Smith 2015), although the mosquito infectome has been studied little. The identification of differences in protein expression between mammalian and mosquito hosts and the functional roles of selected proteins during infection will contribute to the knowledge on factors that affect transmission and epidemic progression.
Ae. aegypti midguts that were infected with CHIKV or DENV-2 viruses were analyzed to evaluate differential protein expression 7 days post-infection (dpi). Gel profile comparisons showed that 8 proteins were modulated by DENV-2, 12 proteins were modulated by CHIKV, and 20 proteins were regulated by both viruses in either similar or different ways (Tchankouo-Nguetcheu et al. 2010). Both viruses caused an upregulation of proteins that were involved in the generation of reactive oxygen species, energy generation, and carbohydrate and lipid metabolism. Midgut infection by DENV-2 and CHIKV triggered an antioxidant response, but CHIKV infection produced an increase in the expression of proteins involved in detoxification. One limitation of this comparative study is that the EIP is different between DENV (10–14 days) and CHIKV (7 days). In another study, CHIKV infection was evaluated in the SG of Ae. aegypti mosquitoes after 3 and 5 dpi. (Tchankouo-Nguetcheu et al. 2012). The authors reported differential expression of 13 proteins at 3 dpi, 19 proteins at 5 dpi, and 10 proteins at both time points during CHIKV infection. The modulated proteins functioned as housekeeping proteins, protease inhibitors, immunity-related proteins and in blood feeding. Recently, Lee and Chu (2015) described the proteome of CHIKV-infected C6/36 Ae. albopictus-derived cells. The authors found 23 cellular proteins that were differentially regulated at different times of infection (1, 2 and 4 dpi). These proteins are involved in diverse biological pathways, including protein folding and metabolic processes, and the most upregulated proteins were the spermatogenesis-associated factor, enolase phosphatase e-1 and chaperonin-60kD. These genes were knocked down with small interfering RNA (siRNA), and interestingly, the results showed a reduction in CHIV infection. These findings have provided insight into the relevance of mosquito host factors in the replication of CHIKV. Protein profiles of Ae. albopictus infected with DENV-2 were also evaluated in the SG, midgut, and in C6/36 cells. The results of this investigation indicate that most of the down- or upregulated proteins are chaperones, cytoskeleton proteins, and energy metabolism enzymes (Zhang et al. 2013). Thus, the SG proteome of Ae. aegypti (Rockefeller strain) infected with DENV2 was analyzed, and the authors reported 7 upregulated and 22 downregulated proteins that were classified as structural, secreted, and metabolic proteins (Chisenhall et al. 2014a). Another study was performed to evaluate the differential proteome in the saliva of Ae. aegypti infected with DENV-2, and the authors described changes in the expression of 23 salivary proteins, particularly proteins that were involved in antihemostatic and pain-reducing capacities (Chisenhall et al. 2014b). These proteins may confer a fitness benefit upon the virus by improving viral establishment in the vertebrate host or by increasing the number of transmission events. Interestingly, the functional analysis of two downregulated SG proteins, namely aegyptin and D7 (Chisenhall, 2014b), indicated that they affected virus replication. McCracken et al. (2014) showed that mice that received aegyptin exhibited decreased DENV titers and an increased immune response when compared to mice that received an inoculation of DENV alone. Additionally, D7 proteins are the most abundant proteins in the mosquito SG (Oktarianti et al. 2015); however, during DENV infection, the expression of D7 is decreased. Conway et al. (2016) revealed that the recombinant D7 protein prevented DENV2 cell binding and/or infection in two permissive cell types and prevented infection and dissemination in a mouse model. These data support the previous observation that D7 protein vaccination enhanced mortality in a West Nile virus mouse model (Reagan et al. 2012).
Based on the results described above, we analyzed the protein network in the midgut or the SGs of Aedes mosquitoes infected with DENV or CHIKV (Figs. 1 and 2). The protein pathway was determined using the STRING and KEGG (Kyoto Encyclopedia of Genes and Genomes) databases. We found that proteomic studies that analyzed vector interactions with DENV or CHIKV uncovered some common patterns in the midgut and SG, such as proteins that were involved in carbohydrate metabolism and the Krebs cycle. Interestingly, the analysis showed that viral infection alters the expression of proteins that are involved in energy metabolism and suggests that infection of the Aedes mosquito induces changes in mitochondrial homeostasis.
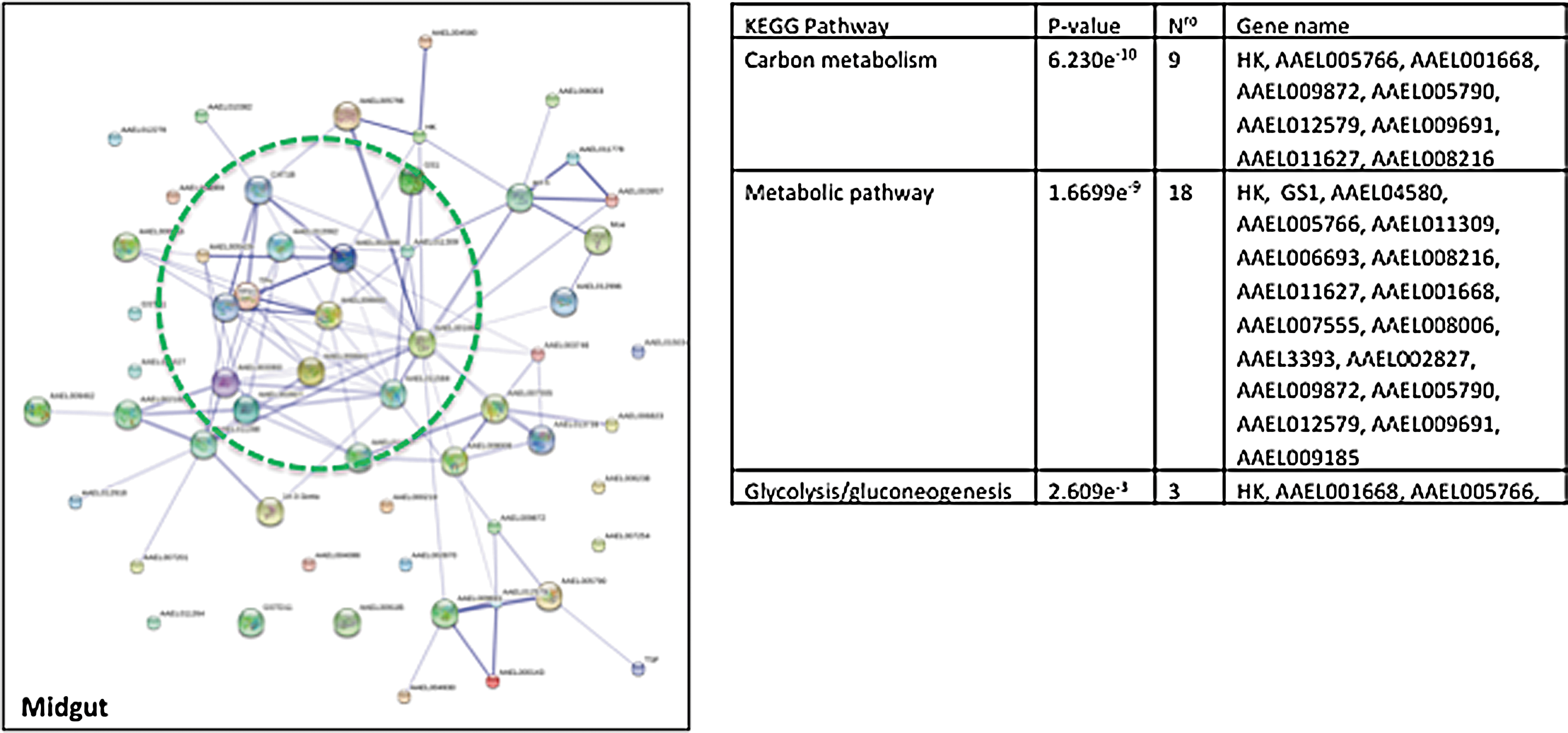
Networks of interactions of differentially expressed proteins that are associated with virus infection in the midgut of the Aedes mosquito. The differential proteins that were reported in the studies cited in Table 2 were used in the STRING analysis at a confidence level 0.7.
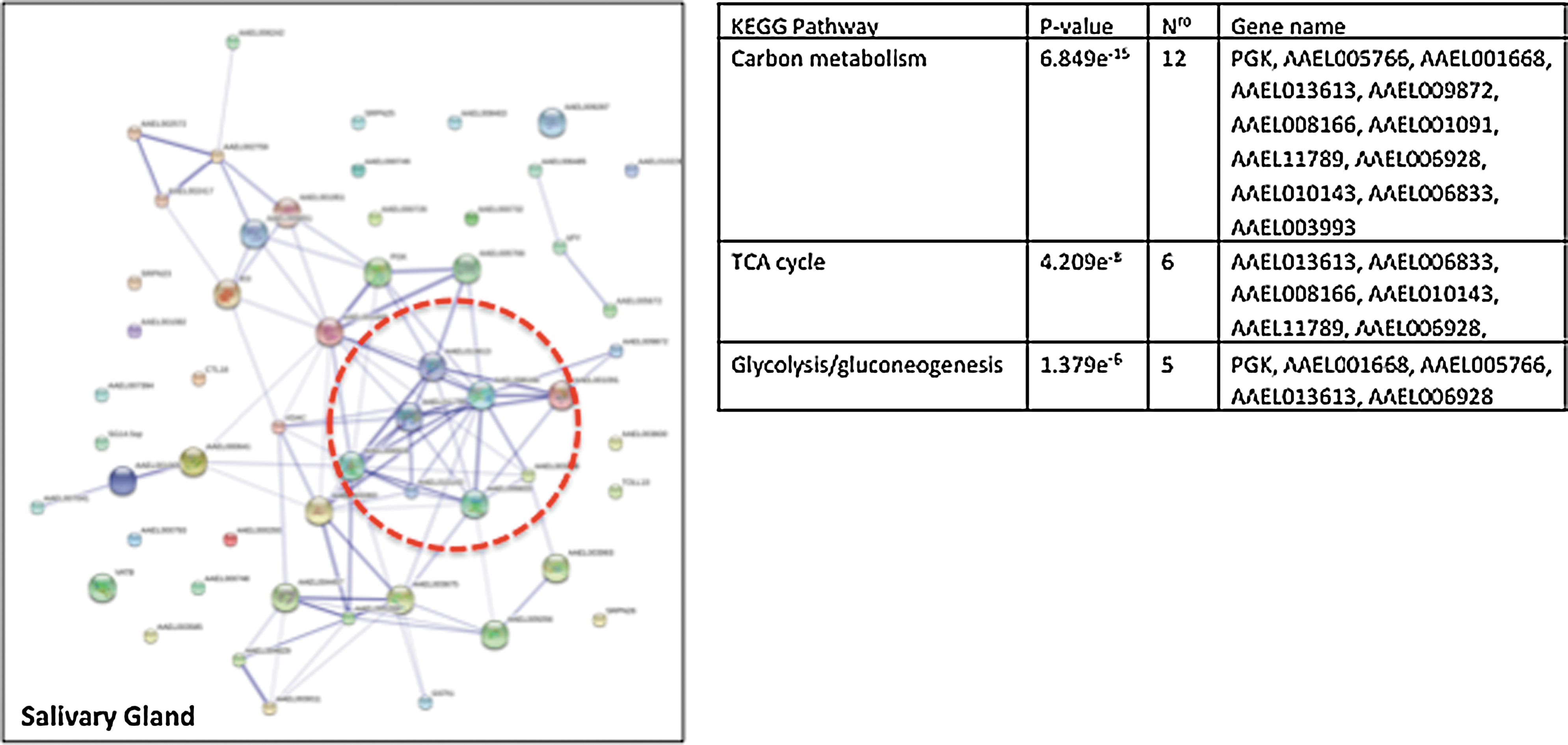
Salivary gland protein networks in the Aedes mosquito infected with DENV or CHIKV. This figure depicts the interactome network of proteins that are modulated by viral infection in mosquito salivary gland tissue as shown in Table 2.
Mitochondria play an essential role in the life and death of the cell. Mitochondria are mainly associated with energy metabolism, and recently, they have emerged as key organelles in the preservation of cellular homeostasis, metabolism, aging, innate immunity, apoptosis, and other signaling pathways (Galluzzi et al. 2012). Viruses interfere with mitochondrial pathways; they strategically alter mitochondrial functions to facilitate their proliferation (Khan et al. 2015). Recently, with the use of a proteomic approach, changes in the expression of 155 proteins were identified in hepatic cells after DENV infection. Of these proteins, 16 were mitochondrial proteins involved in the glycolytic pathway, TCA cycle, and stress response (Pando-Robles et al. 2014). Accordingly, it has been shown that DENV infection promotes changes in mitochondrial bioenergetics and in the respiratory properties of hepatic cells, which causes an increase in cellular oxygen consumption and decreased ATP synthesis efficiency (El-Bacha et al. 2007, Nasirudeen et al. 2008). A reduction in the mitochondrial membrane potential and the release of Cyt c were reported in hepatic and monocytic cell lines upon DENV infection (Thepparit et al. 2013, Olagnier et al. 2014). In addition, reactive oxygen species, which are principally formed in the mitochondria, increase in number during infection (Olagnier et al. 2014). More recently, it has been reported that DENV infection causes an imbalance in mitochondrial dynamics. Yu et al. (2015) found that the DENV protease NS2B cleaved mitofusins (MFN1 and MFN2) and impaired mitochondrial fusion modulating antiviral response and apoptosis. However, Chatel-Chaix et al. (2016) showed that the NS4B viral protein inhibited mitochondrial fragmentation and elongated mitochondria promotes DENV replication and modulates interferon response. These contradictory results indicate that the antiviral response could be cell specific, and it is remarkable that mitochondrial physiology in the invertebrate host during virus infection has not been explored. We highlight the importance of investigations into this particular subproteome.
Proteomics to identify post-translational modifications
The Ae. aegypti genome codes for ∼16,000 genes; however, the total number of functional proteins that can be derived from the genome is not known. Alternative splicing of transcripts is known to increase the protein coding capacity of eukaryotic genomes through the potential generation of more than one messenger RNA (mRNA) from a single gene. Each of these mRNAs is subsequently translated into protein in the cytoplasm. In addition, the number of functional proteins increases through post-translational modifications that modify the chemical composition and structure of proteins, which provides them with further functionalities (Fig. 3). These PTMs can regulate protein activity, localization, and interaction with other molecules such as lipids, nucleic acids, metabolites and other proteins.
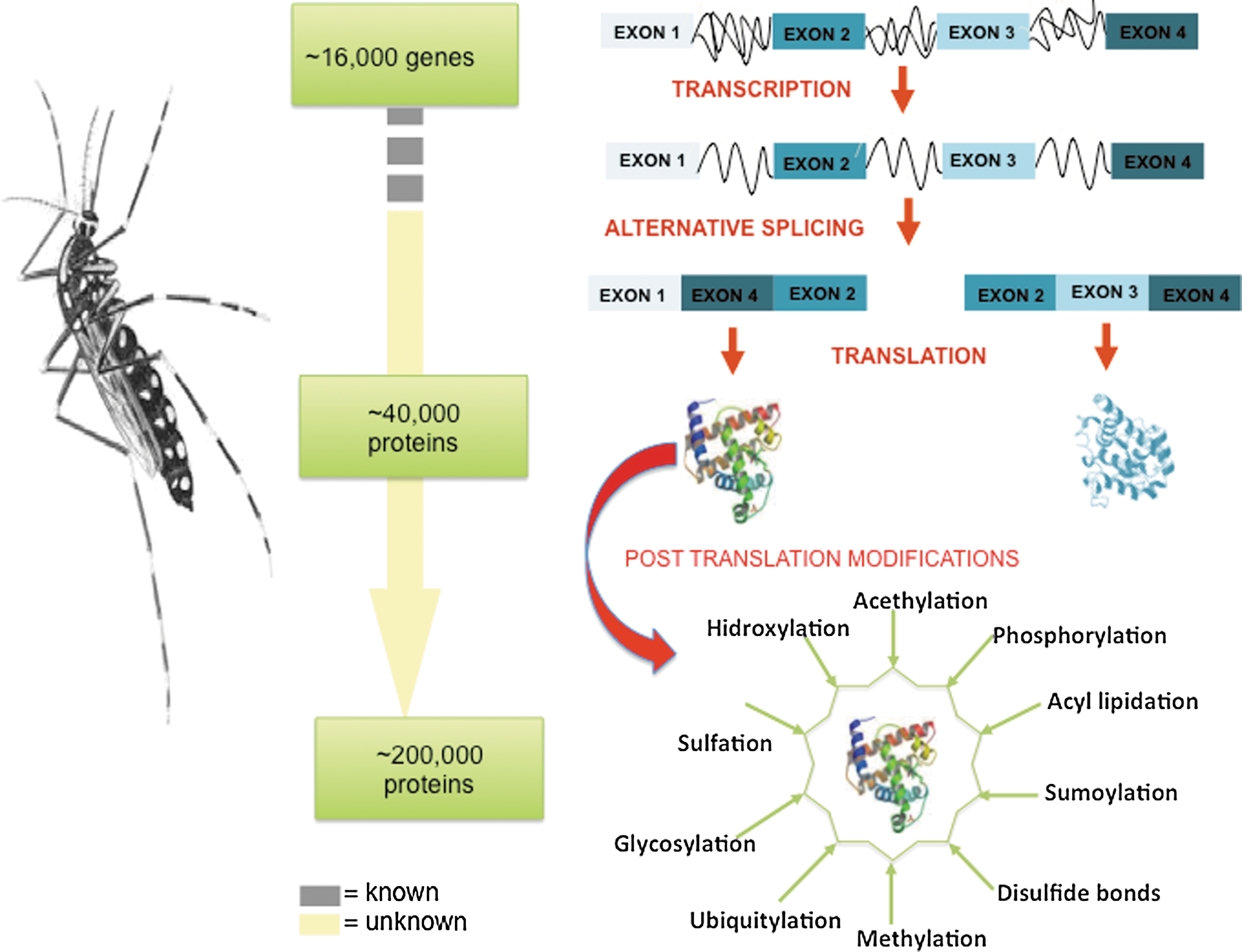
Post-translational modifications are key mechanisms for the expansion of protein diversity. The genome of the Aedes mosquito consists of ∼16,000 genes; the transcription of these genes increases the magnitude of the transcriptome relative to the genome, and the myriad of different post-translational modifications exponentially increases the complexity of the proteome and the functionality of the proteins.
The comprehensive analysis of PTMs on proteins of the Aedes mosquito is crucial to elucidate the regulatory mechanisms of biological processes in the vector and their interaction with the pathogens that mosquitoes transmit to humans. The identification of PTMs has made great progress in the past decade. Specific PTM enrichment techniques have been developed, such as an efficient multidimensional separation strategy. Tools for computational analysis that allow the study of most representative PTMs including phosphorylation, glycosylation, ubiquitination, sumoylation, acetylation, and methylation, among others, have also been developed (Olsen and Mann 2013, Doll and Burlingame 2015, Schwammle et al. 2015).
Protein phosphorylation is the most studied PTM and plays a fundamental role in the regulation of many biological processes. High-throughput phosphorylation analyses have not yet been performed in vector mosquitoes. However, a phosphoproteome of Drosophila KC 167 cells was previously reported (Bodenmiller et al. 2007). This study described 10,000 unique phosphorylation sites that mapped to 4600 distinct phosphoproteins that were encoded by 3500 genes. This information was deposited in Phosphopep, a database for phosphopeptides and phosphoproteins from Drosophila melanogaster. The potential enrichment of this database with information from PTMs in arthropod disease vectors will certainly provide further insights into virus–vector interactions.
Another important PTM that increases the diversity of the proteome is glycosylation. Glycoproteins are one of the major components of viruses and are involved in host specificity, cell tropism, interspecies transmission, pathogenesis, and the immune response (Idris et al. 2016). The DENV surface contains an envelope glycoprotein E with two potential glycosylation sites, Asn 67 and Asn153 (Perera and Khun 2008). However, the types of glycans that attach glycoprotein E remain elusive. Recently, five types of N-glycans were identified on protein E of DENV-2 replicated in insect C6/36 cells; these included mannose, GalNAc, GlcNAc, fucose, and sialic acid, which demonstrates the high heterogeneity of DENV glycans on the viral surface (Lei et al. 2015). Enveloped viral glycoproteins are essential for interactions with host cell receptors and are the main target for the development of vaccines. For this reason, the characterization of protein E glycans is primordial to our understanding of the mechanisms involved in virus entry and on the stimulation of the immune response. Moreover, the CHIKV envelope is composed of glycoproteins E1 and E2. Recently, a strong host-dependent N-glycosylation profile for chikungunya virus-like particles (CHIKV-VLPs) was described (Lancaster et al. 2016). The glycosylation of viral proteins E1 and E2 from CHIKV-VLPs derived from mammalian HEK293 cells included oligomannose, hybrid, and complex glycans. VLPs that originated in insect cells Sf (Spodoptera frugiperda) predominantly contained oligomannose. These differences can be used as valuable tools to evaluate the correlation between immunogenicity and glycosylation. Furthermore, the treatment of cells with certain glycosidases reduced the binding of DENV to mammalian cells but not to mosquito cells (Thaisomboonsuk et al. 2005). Thus, Aedes mosquitoes were recently shown to have the biosynthetic capacity for endogenous sialic acid production (Cime-Castillo et al. 2015). These authors reported that the glycoproteins that were present in mosquito saliva could potentiate virus transmission and suggested that sialic acid modification of mosquito proteins was involved in DENV entry in mammalian cells. Unfortunately, the glycosylation pattern of arbovirus proteins and the carbohydrate profile of the host cell remain incomplete.
On the other hand, ubiquitination is a PTM that is responsible for protein degradation in the cells during normal or pathological conditions. A few years ago, a bioinformatics analysis provided evidence of the presence of ubiquitin machinery in the genomes of five disease vectors, including Ae. aegypti (Choy et al. 2013). In addition, different studies have reported that the ubiquitin proteasome pathway (UPP) is modulated during DENV and CHIKV replication in mammalian cells (Kanlaya et al. 2010, Fischl and Bartenschlager 2011, Morrison et al. 2013, Karpe et al. 2016). The DENV NS5 protein inhibits interferon signaling by mediating proteasome-dependent STAT2 degradation (Morrison et al. 2013). Additionally, during CHIKV infection, the accumulation of polyubiquitinated proteins, which is induced by proteasome inhibition, and the earlier visualization of the UPR has been reported (Karpe et al. 2016). Recently, the UPP was shown to be critical for infectious DENV production in the mosquito midgut. Downregulation of the catalytic subunits of the proteasome, namely β1 (caspase-like activity), β2 (trypsin-like activity), and β5 (chymotrypsin-like activity), reduced viral progeny production but did not influence viral genome replication (Choy et al. 2015). This information suggests that an analysis of the ubiquitination of proteins may provide important clues to the regulation of the mosquito response to viral infection.
Peptidomics
As components of signaling regulatory networks, peptides also play an important role in many cellular functions. Therefore, it is imperative to characterize the peptide composition of mosquito tissues and to elucidate their role in mosquito biology and virus–vector interactions. Neuropeptides represent the largest single class of signal compounds that are involved in the regulation of development, growth, reproduction, metabolism, and behavior of insects (Altstein and Nässel 2010). Endocrine cells and neurons produce neuropeptides and protein hormones as larger precursors (prepropeptides), which are processed, stored and released within the nervous system as neurotransmitters or neuromodulators. Based on information from annotated genomes of several species of insects including Ae. aegypti, it is known that there are approximately 30–40 genes that encode neuropeptide precursors (Hummon et al. 2006). Furthermore, Predel et al. (2010) performed a comprehensive peptidomic characterization of neuropeptides from Ae. aegypti via MS and identified 80 neuropeptides of selected tissues from a single specimen analysis. This study provides a framework for future investigations on mosquito endocrinology and neurobiology, especially in the context of viral infection.
Lipidomics
The architecture of biological membranes is dependent on the composition and distribution of lipids and proteins. The replication of the majority of RNA viruses is associated with intracellular membranes and results in a dramatic reorganization of membrane architecture in the cell during virus infection. Flavivirus replication induces invaginations in the ER membranes and forms vesicles where RNA replication occurs (Welsch et al. 2009). Similarly, alphavirus induces small invaginations called “cytopathic vacuoles” (CPVs) in the plasma membrane, which are internalized and become part of the endo-lysosomal membrane system and give rise to CPVs where RNA replication occurs (Paul and Bartenschlager 2013). These specialized membrane compartments can assist in the evasion of host antiviral defense mechanisms and also function to increase the local concentration of molecules that are necessary for efficient viral RNA replication and virus particle assembly (Welsch et al. 2009, Diaz et al. 2010). The dysregulation of lipid metabolism has been implicated in some diseases, including viral infections such as dengue and chikungunya. The redistribution of cellular lipids to replication sites appears to favor virus replication and survival, but the nature and specificity of this phenomenon are not well understood. To extend these studies, high-resolution MS was recently used to evaluate global changes in lipid metabolism upon DENV infection (Perera et al. 2012). The authors found drastic alterations in the global lipid profile in both human (hepatic cells, Huh-7) and mosquito (Ae. albopictus derived C6/36 cells) cells upon DENV infection. Furthermore, Barletta et al. (2016) reported that genes that were associated with lipid droplets, biogenesis, and lipid metabolism were regulated after bacterial or dengue infection in the mosquito cells, which suggests that lipid droplets have a role in mounting an effective antiviral response. In addition, the function of two lipid-binding proteins, the myeloid differentiation 2-related lipid recognition protein (ML) and the Niemann Pick-type C1 (NPC1) families, were analyzed during DENV infection in the mosquito. The depletion of these proteins causes an inhibition of virus production in mosquito midguts, which suggests that the expression of these proteins facilitates DENV infection of the mosquito host (Jupatanakul et al. 2014). Knowledge of how the DENV utilizes cellular lipid metabolism and downstream signaling pathways to facilitate its replication will provide novel targets that could be useful for the development of effective antivirals for vector control.
The challenge of studying mosquito proteins
Proteomics is one of the fastest developing areas of research and offers insight into the biological processes and signal pathways that change in the cell as a consequence of virus infection. Although significant advances in the comprehensive profiling, functional analysis, and regulation of proteins have occurred in invertebrate model organisms such as D. melanogaster, proteomics research in mosquito vectors has not advanced at the same pace. Recently, with the availability of the complete Ae. aegypti and Ae. albopictus genomes, it is possible to conduct large-scale proteomics studies. However, there are two major obstacles to the characterization of vector–virus interactions at the level of proteins and their PTMs. First is the limited amount of biological material that can be obtained from a single mosquito. Second, but more important, is the distinct dynamics of viral infection for each particular mosquito tissue. It is important to point out that, in contrast to transcriptomics, there is no signal amplification technique, such as PCR, for proteomics studies. Therefore, proteomics studies have to rely on the level of a raw signal from the protein composition of samples. With proteomics, researchers can improve gene annotations, especially for sets of genes with characteristics that make them difficult to predict with gene-finding algorithms. MS data can also used to identify new genes in a specific tissue or biological process, particularly on the genome of mosquito Aedes that has a high percentage of transposable regions. To overcome these hurdles, communication among entomologists, virologists, and spectrometrists is critical to advance this field and to contribute to the prevention and control of Aedes-borne viral infections.
Concluding Remarks
Arboviruses cycle between insects and vertebrate hosts; due to this alternation, they are obligated to use different cellular machinery for their replication, and the outcome of infection is dependent on the host. The involvement of host factors in viral replication and viral protein synthesis is an area that is only just beginning to be explored and will likely improve our understanding of virus species-specific pathogenesis. In addition to the pending studies that are proposed above and the application of newer techniques to further dissect virus–vector interactions, there are some issues that will be relevant to the understanding of such interactions. We suggest at least three, and these are as follows: (1) Aedes females have a high demand for energy during virus infection, and ATP is generated at the mitochondria in different insect cells. However, the role of energy metabolism and the role of reactive oxygen species have not been investigated. We suggest that it is important to investigate this particular subproteome. (2) Post-translational modifications provide proteins with additional properties that render them potentially multifunctional. However, the potential role of PTMs in the vector–virus interaction has not been investigated in Aedes. Current proteomics methods could be applied to characterize the phosphorylation and ubiquitination of mosquito proteins at a high-throughput level. These two types of modifications would be of particular interest because they could be involved in the Aedes immune response to viral infections. (3) Although the expression of many proteins is known to change in response to viral infections in the mosquito, the corresponding functional studies on the role of these proteins in viral infection are still pending in the majority of cases.
Footnotes
Acknowledgments
This work was partially financed by CONACYT grant 179865 and UCMexus 2015 to V.P.R. This article was edited for English language by American Journal Experts (AJE).
Authors' Contributions
V.P.R. conceived the article and wrote the initial draft. C.V.F.B. and V.P.R. edited subsequent versions of the article and approved the final version.
Author Disclosure Statement
No competing financial interests exist.