
Abstract
Toxoplasma gondii is a protozoan parasite with a broad ecological valence, which has been detected in a wide range of hosts and landscapes. Although the genus is considered monospecific, in recent years it has been demonstrated to exhibit more genetic variability than previously known. In Mexico, there are few genotyping studies, which suggest that classical, autochthonous, and atypical strains are circulating. The goal of this study was to describe T. gondii genetic diversity in naturally infected sheep from Colima, Mexico. This is a good site to study ecological aspects of this parasite since it is located between the Nearctic and Neotropical ecozones and it includes domestic and wild risks for transmission. We analyzed 305 tissue samples of semicaptive sheep from six coastal and central zones of Colima and border zones of Michoacán. We used an 803 bp amplicon of the B1 gene to genotype T. gondii and seroprevalence was determined by ELISA. Indexes for genetic diversity and genetic differentiation were calculated and compared with reference strains from North America (NA) and South America (SA). Twenty-three tissue samples were positive for the B1 gene by PCR, which were sequenced. Crude prevalence was 24.4%. The genetic analysis showed 16 variable sites along the 803 bp region that grouped all sequences into 13 haplotypes in the phylogenetic tree. Bayesian and haplotype network analysis showed nine new B1-types, of which three were frequent and six had unique alleles. Comparisons among sequence sets revealed that the Mexican population had lower differentiation than SA and an intermediate genetic variability between South America and North America. The B1 gene analysis showed new T. gondii haplotypes in naturally infected sheep; therefore, this marker could be initially used in molecular screening studies to identify potentially virulent genotypes of this parasite using natural host samples directly.
Introduction
T
In consequence, it has been argued that different parasite populations are predominant in the sylvatic and domestic cycles, but which can traverse from one to the other, like in other emerging infectious diseases (Ambroise-Thomas 2000, Daszak 2000, Robertson et al. 2000, Polley 2005, Roche and Guégan 2011, Wendte et al. 2011, Grogan et al. 2014).
The genetic diversity of T. gondii has not been well studied in Mexico, although some reports show the presence of archetypal and atypical genotypes (Dubey et al. 2004, 2009, 2013, Cedillo-Peláez et al. 2011, Rico-Torres et al. 2012, 2015). As an example, a strain isolated from a wild puma (Felis concolor) from northern Mexico was typed as ToxoDB #222, a new atypical genotype, virulent for mice (Dubey et al. 2013). Epidemiological surveys in human populations have revealed toxoplasmosis to be endemic in the whole country, being higher in the subtropical areas (Velasco-Castrejón et al. 1992, Caballero-Ortega et al. 2012). Specifically, in the Pacific Coast, the Gulf of Mexico and the Yucatan Peninsula, the human infection exceeds 70% (Caballero-Ortega et al. 2012).
It also has been reported that exposure to T. gondii displays a stratified prevalence in sheep flocks, being higher in lowlands than in highlands (Caballero-Ortega et al. 2008). These patterns may be of global relevance since biogeographically this region borders a transitional area between the Nearctic and Neotropical ecozones (Fosberg 1976, Olson and Dinerstein 1998, Olson et al. 2001, Morrone 2015), which has been related to differential T. gondii distribution and genetic diversity (Khan et al. 2007, Su et al. 2012).
The division caused by the mountain ranges represent physical and ecological barriers that isolate natural populations at both sides (Morrone 2015), which has let a great variety of potential intermediate hosts, including the six species of wild felids (Ceballos and Oliva 2005), as well as, domestic cats freely roaming in the rural, semiurban, and urban areas (García-Márquez et al. 2007, Caballero-Ortega et al. 2008, Rico-Torres et al. 2015). In light of these findings, the aim of this study was to describe the genetic diversity of T. gondii in a pacific coastal region of Mexico in semicaptive sheep by direct DNA analysis using the B1 gene, due to its effectiveness to identify virulent or new genotypes (Grigg and Boothroyd 2001, VanWormer et al. 2014).
Materials and Methods
The present study was approved by the Use of Laboratory Animals Care and the Research Committees of the Instituto Nacional de Pediatría (reference number 013/2012).
Sample collection
Small pieces of muscle tissue from the leg, diaphragm, and heart, as well as ∼20 mL of peripheral blood for serology screening, were collected from March 2013 to March 2014 from the municipal slaughterhouse of Colima, according to the Mexican official standard NOM-033-ZOO-1995. All biological samples belong to semicaptive animals destined for human consumption; therefore, no sheep was euthanized for this study. Fresh tissue samples were washed in a commercial solution of 1% disinfectant (VIRKON S, BAYER, Mexico) and placed in individual bags to avoid cross contamination. Sample sets were transported by air cargo within 24 h postcollection and carried in cold chain until DNA extraction was made in the laboratory in Mexico City. Metadata for the animals (age, sex, and flock location) were recorded during sample collection. Serum samples were tested by a previously standardized indirect ELISA (Caballero-Ortega et al. 2008).
DNA extraction
Before DNA extraction, samples were processed with a tissue cyst concentration technique based on density gradient centrifugation by sucrose surface flotation. Briefly, 5 g of pooled tissues were minced with a hand blender in 40 mL of bidistilled water for 2 min. Homogenates were filtered through a sieve with 180 μm opening (Newark Wire Cloth Company, NJ, USA) and poured into 50-mL conical centrifuge tubes.
After filtration, two 5-min washing steps at 3000 g were made with bidistilled water. The pellet was resuspended in a sucrose solution (specific gravity = 1.2); volume was adjusted to 50 mL and centrifuged at 1500 g for 3 min. Five hundred microliters of the supernatant were transferred to 1.5-mL microcentrifuge tubes to make two washings with bidistilled water at 184,800 g for 10 min. Resulting pellets were subjected to DNA extraction using a protocol to process small samples with organic solvents (Green and Sambrook 2012). DNA was dissolved in 50 μL DNA hydration solution (QIAGEN, Germany), incubated at 65°C for 1 h, and stored at −20°C.
Genetic diversity and phylogenetic analysis
Direct DNA amplification was performed by established nested-PCR protocols, PCR-screening for T. gondii infection and genotyping was done by endpoint PCR of 362 bp (Rico-Torres et al. 2012) and ∼500 bp (Grigg and Boothroyd 2001) fragments, both of the B1 locus. In addition, haplogroup identification was attempted by PCR sequencing of intronic regions with a set of eight unlinked markers (Khan et al. 2007).
T. gondii DNA of RH strain was used as positive controls. PCR amplification was carried out in 25 μL mixes and 35 cycles for the external primers, and 100 μL with 25 cycles for internal primers in the nested PCR. For all set of primers, concentration of reagents was calculated for 100 μL volume reaction and were as follows: 1 × PCR buffer, 1.5 mM MgCl2, 25 pmol of each primer, 1 × enhancer buffer with Betaine (Promega, USA); BSA 0.4% (vol/vol); and 200 μM of each dNTP and 3 U of Taq polymerase. Template DNA ranged from 200 ng to 1.0 μg for each pair of primers. PCR products were purified with the QIAquick Gel Extraction Kit (QIAGEN, Germany) and sequenced by a commercial supplier.
To analyze the polymorphic sites, chromatograms were evaluated with the Mesquite software (Maddison and Maddison 2016b) using the Chromaseq package (Maddison and Maddison 2016a) that partially makes use of the Phred and Phrap algorithms for base calling, assigning quality values to each called base, and assembling contigs (Ewing and Green 1998, Ewing et al. 1998, Gordon et al. 1998). BLASTN searches were run using the complete sequence of the B1 locus reported for the RH strain (Burg et al. 1989) as the query (GenBank: AF179871.1) against the Whole-Genome Shotgun (WGS) Contigs projects from the 16 T. gondii genomes hosted at the NCBI.
Sequence alignments were made with ClustalW version 2.1 (Larkin et al. 2007). Phylogenetic reconstruction was computed with MrBayes Software version 3.2.6 (Ronquist et al. 2012). Sequence data were partitioned by intron and exon, analysis was performed for 10 million generations, posterior probability distribution and diagnostic frequency were sampled every 1000 and 100,000 generations, respectively, substitution model was implemented during analysis by reversible jump, trees were summarized with a postburnin sample of 50% and modified with Figtree v1.4.2 (“FigTree,” n.d.). Unrooted haplotype networks were computed using NETWORK version 4.611 (“
DNA polymorphism analysis was performed in DnaSP v5.10.01, which included estimation of nucleotide diversity (π), haplotype diversity (Θ), gene flow (Nm), and genetic differentiation (HST). Permutation test was computed to define significance levels (Librado and Rozas 2009). The common values used for genetic differentiation were: 0–0.05, small; 0.05–0.15, moderate; 0.15–0.25, great; values above 0.25 indicate huge genetic differentiation. The gene flow or migration index (Nm) refers to the movement of organisms among subpopulations; those strongly differentiated have an Nm << 1, whereas an Nm >4 behaves as a single panmictic unit (Hartl and Clark 2007).
Results
We obtained tissue samples from 305 semicaptive sheep destined for human consumption during 1 year, from 27 locations of Colima and two of Michoacán, Mexico. Based on their distance from the coastal line and the geographical location, sampling zones were grouped into six sites (S). Sites S1 to S4 and S6 are located ∼2, ∼14, ∼13, ∼54, and ∼29 km away, respectively, from the coastal line in the state of Colima, whereas S5 was located ∼10 km away from the coastal line in a border area between both states (Fig. 1).
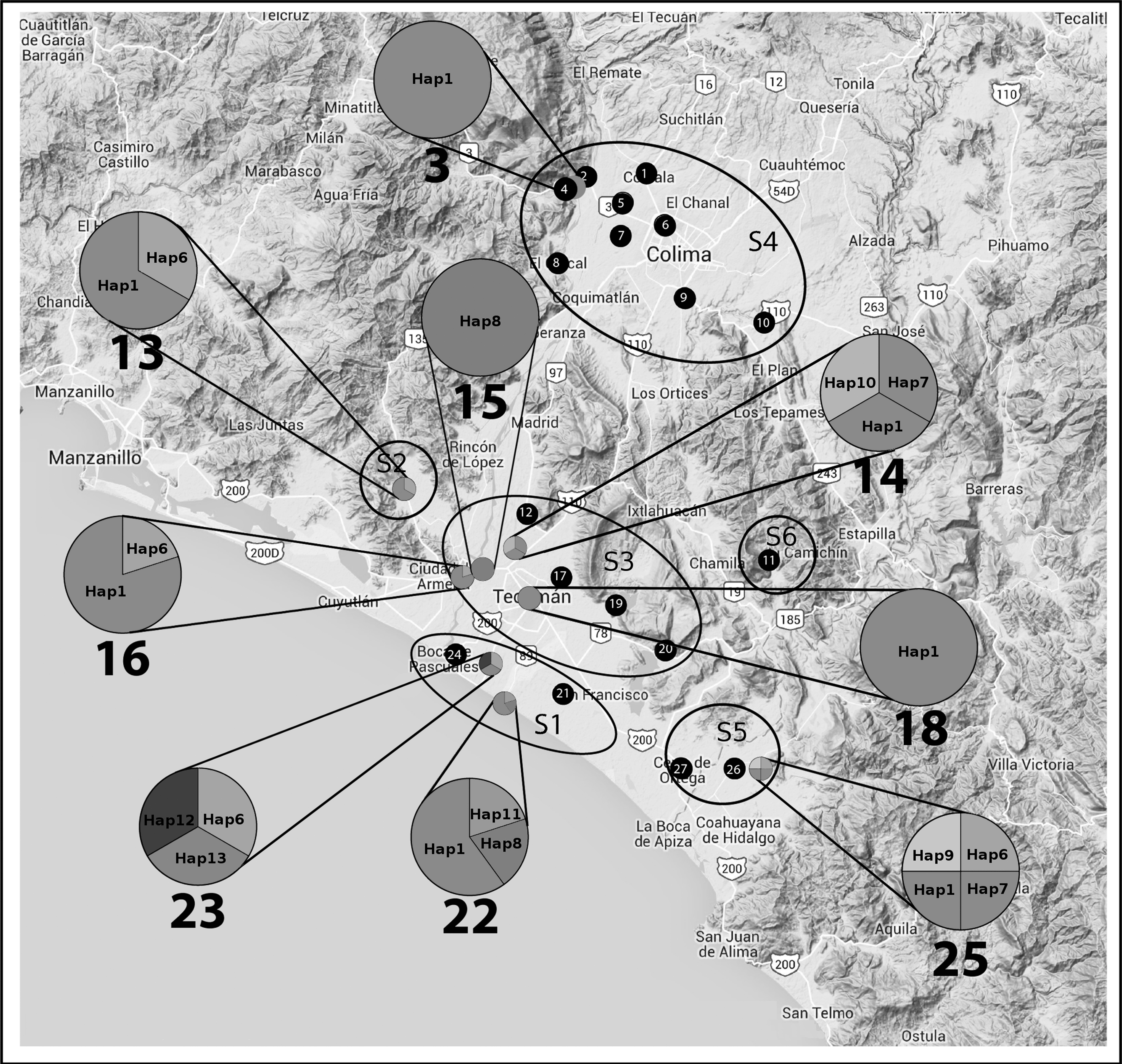
Distribution of locations and haplotypes of Toxoplasma gondii in a Pacific coastal region from Mexico. Sampled locations were positioned by its geographic coordinates and grouped into S1 to S6 by its proximity. Locations in which samples could be sequenced are represented with pie charts that match haplotype frequency, whereas solid black charts represent sampled locations from where no sequences could be gathered. Enlarged pie chart size was normalized for a better view.
From the whole set of samples, 275 were obtained from S1, S2, S3, and S5, whereas 30 were gathered from S4 and S6; 64% (n = 196) were males, 32% (n = 99) females, and 4% (n = 10) fetuses of undefined sex. Four age groups were established: 6% fetuses, 68% <1 year, 15%> 2 and <4 years, and 11%> 4 years. The frequency of antibodies was 24.4%, like in a previous study in the same area, 9 years ago (Caballero-Ortega et al. 2008). From the total DNA set of samples, 23 were positive for the multicopy B1 locus in both contiguous regions of ∼500 and 362 bp; therefore, concatenated sequences of 803 bp were analyzed (Supplementary Table S1; Supplementary Data are available online at
Chromatogram analysis in both forward and reverse sequence trace files from each sample, revealed high-quality scores, good intensities, and well-defined peaks with no background noise in the position of interest, showing new sequence haplotypes for 13 samples, of which, three sequences uncovered unresolved nucleotides (Estacion101 and Camalote102 position 305 C/t; ElReal109 position 754 A/g).
Samples with new haplotypes were resequenced at least three times; the pattern and quality remained in chromatograms, even in those with unresolved nucleotide positions for three samples, in which two sequences (A and B) were generated. Sequences obtained were submitted to the GenBank database and are publicly available with accession numbers KX270363–KX270388. No useful DNA products or sequences were obtained with any of the eight unlinked markers used to type haplogroups. During the BLAST searches against NCBI nucleotide databases, we found whole B1 gene sequences for 16 different strains; detailed information is provided in the Supplementary Table S2. No manual edition was needed for the DNA alignments.
The haplotype network tree showed the existence of 16 variable sites along the 803 bp region that grouped the 40 sequences into 13 haplotypes (Fig. 2). Polymorphic sites were positioned within the 2214 bp sequence tandem of the B1 locus; only polymorphic sites 504 (G/c) and 754 (A/g) of the original report documented by (Burg et al. 1989), were identified in the present study. All sequences from sheep were similar to type I alleles, but new single nucleotide polymorphisms at different sites were recognized at position 172 (C/t) for the Hap6 (in four samples), 305 (C/t) for the Hap7 (in two samples), and 679 (C/t) for the Hap8 (in two samples). Sequences of the reference strains were clustered within Hap1–Hap5, whereas those obtained in the present study were grouped within Hap1, as well as in Hap6–Hap13 (Supplementary Table S1).
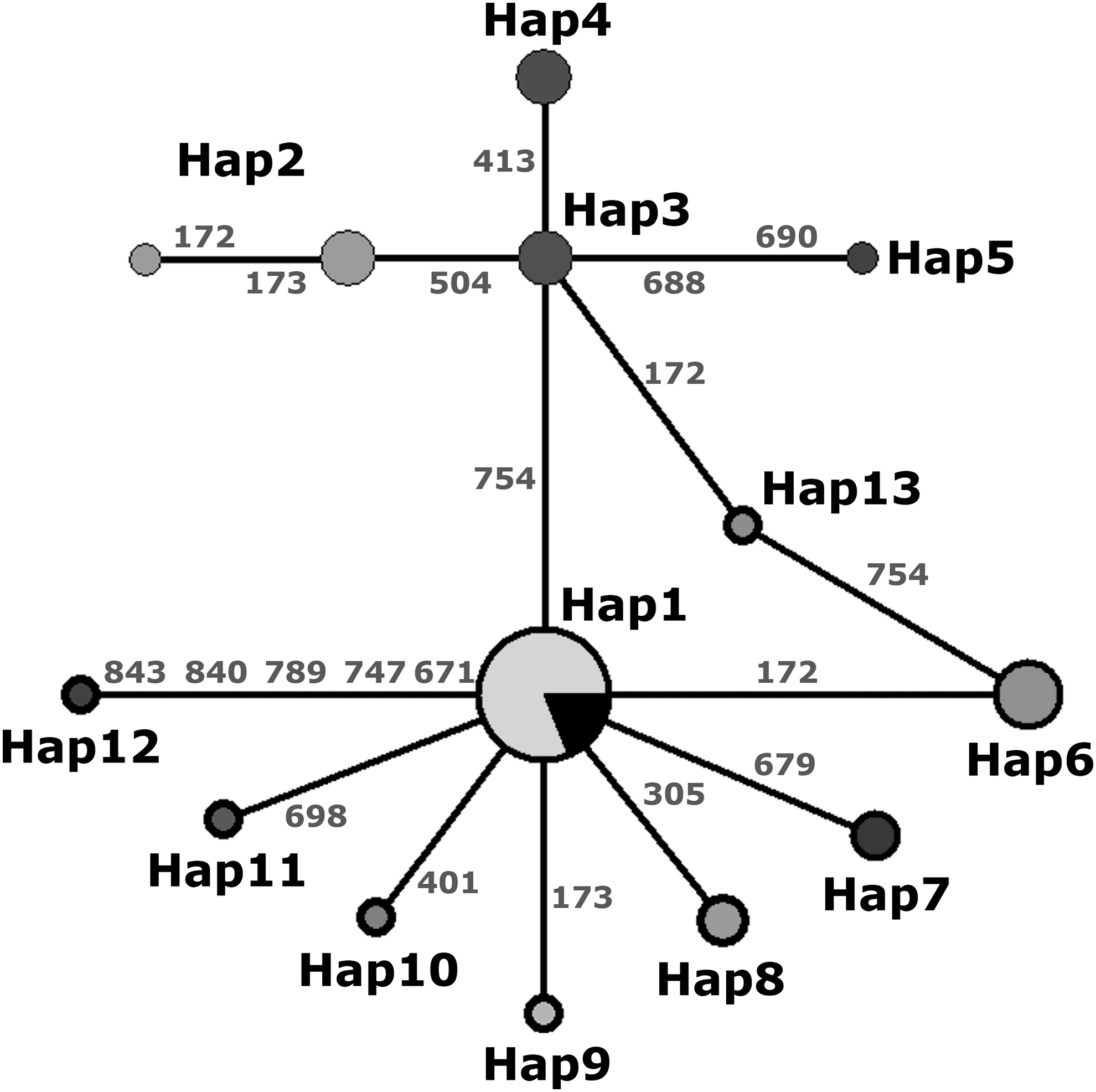
Haplotype network of the B1 gene sequences. Pie chart sizes are proportional to haplotype frequencies. Haplotypes (Hap) identified in the present study are represented by pie charts with bold outline, whereas those from online databases are delimited by thin lines. The black portion in the type 1 chart indicates those sequences from online databases. Numbers indicate single nucleotide polymorphism positions within the 2214 bp tandem of the B1 gene.
Based on sequence comparisons of the B1 gene from the T. gondii WGS-projects available, we found that the complete 2214 bp region presents 26 polymorphic sites that can distinguish every strain from others, except for TgCatBr5 and MAS strains, which have identical sequences (Supplementary Table S3).
The Bayesian phylogeny (Fig. 3) showed that seventeen of our samples were clustered with sequences that exhibited the type I alleles, including Hap9–Hap12, identified by the network tree as unique haplotypes (Fig. 2). Sequences belonging to haplotypes 6, 7, and 8 from the network tree were also grouped as separate clades in the Bayesian reconstruction. Finally, the sequence corresponding to haplotype 13 was included in another branch that groups most of the nontype I reference strains within independent clusters.
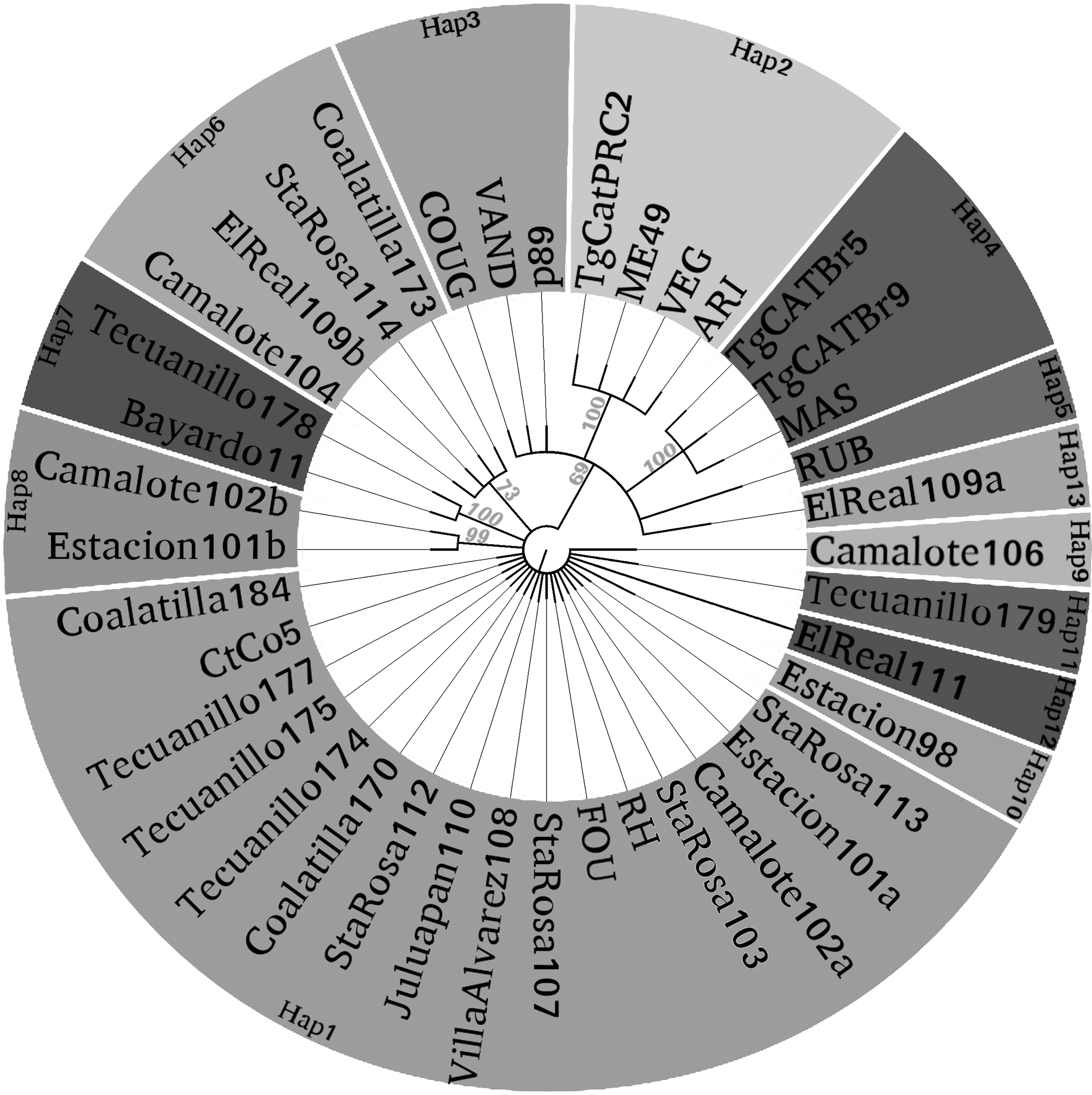
Phylogenetic tree of all sequences with the MrBayes algorithm. Clustering of haplotypes (Hap1 to Hap13) found in semicaptive sheep from Colima and their relationship with reference strain sequences. Numbers indicate support values for each cluster; the reference numbers or name of strains are placed at the end of each branch; bold names represent sequences from this study. Haplotype code names match those in Figures 1 and 2.
Values for HST and Nm indexes showed low levels of genetic differentiation (HST ∼0.05, Nm ∼8) for comparisons between sequences reported here and those from South America (SA) or North America (NA), respectively. Although, nucleotide and haplotype polymorphism values were similar among the three geographical regions (π ∼0.001, Θ ∼0.002), the SA dataset showed higher genetic variability, followed by Colima and NA, respectively (Supplementary Table S4).
Discussion
Knowledge about the genetic structure of parasite populations could be an important tool applied to their control because genetic variation within and among populations could determine evolutionary changes, genetic differentiation, and speciation events (Thompson and Lymbery 1995, Rodriguez-Prado et al. 2014). To increase the knowledge of the genetic diversity of T. gondii, we chose ovines kept under semicaptive conditions from a hyperendemic area in Mexico, because they are good sentinels of environmental contamination with oocysts and may be mediating two-way domestic–wild cycles crossing (Velasco-Castrejón et al. 1992, García-Márquez et al. 2007, Caballero-Ortega et al. 2008, 2012).
Based on Bayesian reconstruction and haplotype network, we found a type I-like gene pool in our samples; however, it was possible to identify new haplotypes by direct DNA analysis of the B1 locus; therefore, our data are complementary to those reports which described the existence of archetypal lineage I infecting hosts in Mexico (Cedillo-Peláez et al. 2011, Rico-Torres et al. 2012).
Since the Nearctic and Neotropical ecozones converge in Mexico, specific settings of geographical and ecological features are present, exhibiting variable climates and biomes. Also, mountain ranges are playing the role of physical barriers that might facilitate the isolation of hosts and consequently parasite populations between these ecozones (de Meeûs et al. 1998, Bozick and Real 2015). In contrast, a high gene flow and scarce differentiation within population of each ecozone were observed; our data of huge gene flow between samples suggest that T. gondii in the Pacific coastal region acts like a metapopulation (ensemble of interacting populations with a finite lifetime) and it implies a link with the processes of population turnover, extinction, and establishment of new populations (Levins 1969, Hanski and Gilpin 1991).
A recent study performed in cats of different locations of Colima also shows the existence of a type I gene pool in this Neotropical ecozone (Rico-Torres et al. 2015). In contrast, studies with animals performed in the northern state of Durango, Mexico, placed in the Nearctic area showed dominance of the clonal Type III lineage (Dubey et al. 2004, 2009).
Although the presence of geographical barriers strengthens the separation of parasite populations between Nearctic and Neotropical ecozones, it is logical to assume that gene flow might be occurring within those available convergence zones. In the neighboring states of Colima and Michoacán, a stratified prevalence on sheep flocks related to the altitude has been found; in the former, rates were higher in lowlands than in highlands, whereas in the latter, an opposite phenomenon was described (Caballero-Ortega et al. 2008, Alvarado-Esquivel et al. 2013), showing the relevance of the local biomes for transmission and epidemiology of this parasite. Furthermore, the role of the definitive host in the transmission of the infection in this area should be emphasized.
In this regard, an epidemiological survey in cats from urban and periurban microzones of Colima City showed that prevalence was mainly related to raw meat consumption, as opposed to commercial food feeding (García-Márquez et al. 2007). In addition, ToxoDB #28 was demonstrated in the first genetically characterized T. gondii isolate from Colima, which was different from others of the country, but similar to those of USA, Brazil, and Colombia (Howe and Sibley 1995, Dubey et al. 2006, 2008, Rico-Torres et al. 2015).
Although our data suggest clonality within population of a given ecozone, when the π, Θ, HST, and Nm indexes were compared with sequences of strains from NA and SA, the results exhibited a lower differentiation between the Pacific coastal region and the SA, than between Colima and NA, suggesting similitude of biomes along the Neotropical ecozone of Central and South America.
An initial plus stated for the present study was to genotype without isolation in mice. As it is known, isolation success in rodents depends on the virulence and parasite load, avirulent strains resulting in chronic infections with a low number and nonhomogeneous tissue cyst distribution, whereas virulent parasites give rise to rapid host death because of a disseminated toxoplasmosis. Thus, this approach could bias isolation to certain strains over others (Lindström et al. 2008, Sibley et al. 2009), and real genetic variability may be underestimated, resulting in descriptions of clonal populations.
To avoid this bias, the first-approach direct genotyping, like the one performed herein, can be used. Before DNA extraction, we attempted to concentrate tissue cysts from host tissue to increase parasite DNA and improve PCR-based results; however, genotyping from direct DNA analysis was rather difficult. These results might be influenced by several factors, such as number, distribution, and age of the tissue cysts, copy number of the loci used and parasite DNA required to sequencing, as well as the age of the sheep analyzed (Lundén et al. 1994, Dubey 1998, Lindström et al. 2008, Sibley et al. 2009, Costa and Bretagne 2012, Robert-Gangneux and Darde 2012).
B1 gene genotyping presents some advantages over those single-copy multilocus approaches due to its simplicity, higher sensitivity, cost effectiveness, and reproducibility, without the need for isolation. However, in comparison with single-copy markers, it lacks genotyping resolution, so results must be interpreted with caution, since sequencing alone cannot differentiate polymorphisms from coinfections (Sibley et al. 2009, Su et al. 2010).
Certainly, there is a limited understanding of how genetic polymorphisms influence virulence or parasite epidemiology in T. gondii; therefore, we propose that B1 gene could be used as a first approach marker for screening prevalent genotypes in new hosts or landscapes. Then, finer multilocus nested PCR-restriction fragment length polymorphism-sequence analysis (Su et al. 2010) can be attempted in conjunction with the methodology to concentrate intramuscular tissue cysts used in the present study.
Conclusion
We identified new genotypes related to the type I lineage of T. gondii by direct DNA analysis using the B1 locus, infecting sheep destined for human consumption in a Pacific coastal region from Mexico. In this area, Nearctic and Neotropical ecozones converge, landscaping to parasite populations as a metapopulation, maintaining some levels of gene flow with populations from South America.
Footnotes
Acknowledgments
This work was supported by INP grant 013/2012 for sampling collection, analysis, and interpretation of the data, as well as writing and publication of this article. Williams Arony Martínez-Flores is a doctoral student from Programa de Doctorado en Ciencias Biomédicas, Universidad Nacional Autónoma de México (UNAM) and received fellowships 245622 and 22961 (CB-2011/168619) from Consejo Nacional de Ciencia y Tecnología (CONACYT).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.