
Abstract
In Australia, infection of horses with the West Nile virus (WNV) or Murray Valley encephalitis virus (MVEV) occasionally results in severe neurological disease that cannot be clinically differentiated. Confirmatory serological tests to detect antibody specific for MVEV or WNV in horses are often hampered by cross-reactive antibodies induced to conserved epitopes on the envelope (E) protein. This study utilized bacterially expressed recombinant antigens derived from domain III of the E protein (rE-DIII) of MVEV and WNV, respectively, to determine whether these subunit antigens provided specific diagnostic markers of infection with these two viruses. When a panel of 130 serum samples, from horses with known flavivirus infection status, was tested in enzyme-linked immunosorbent assay (ELISA) using rE-DIII antigens, a differential diagnosis of MVEV or WNV was achieved for most samples. Time-point samples from horses exposed to flavivirus infection during the 2011 outbreak of equine encephalitis in south-eastern Australia also indicated that the rE-DIII antigens were capable of detecting and differentiating MVEV and WNV infection in convalescent sera with similar sensitivity and specificity to virus neutralization tests and blocking ELISAs. Overall, these results indicate that the rE-DIII is a suitable antigen for use in rapid immunoassays for confirming MVEV and WNV infections in horses in the Australian context and warrant further assessment on sensitive, high-throughput serological platforms such as multiplex immune assays.
Introduction
W
MVEV and WNV activity is monitored in Australia by sero-conversion in sentinel chickens and by virus isolation from captured Culex annulirostris mosquitoes, which is the main transmission vector for these viruses (Mackenzie et al. 1994, Bolisetty et al. 2002). Occasionally, these viruses spread from their northern endemic foci to more southerly regions during times of unusually high rainfall (Mackenzie et al. 1994, Brown et al. 2002, Hall et al. 2002, Mackenzie and Williams 2009).
In 2011, there was an outbreak of equine viral encephalitis in south-eastern Australia with at least 1000 documented cases, presenting with clinical signs consistent with those observed in horses infected with WNV in the United States (Frost et al. 2012, Mann et al. 2013, Speers et al. 2013). The case-fatality rate for this outbreak was 10–15%, and the primary causative agent was found to be a variant strain of WNV (Frost et al. 2012). During this period, several confirmed cases of MVE in horses and humans were also recorded in south-eastern Australia (Speers et al. 2013).
Although viral RNA was detected, using RT-PCR, in brain specimens from animals succumbing to severe clinical symptoms, virus was not isolated from the blood and serum, likely due to the transient, low-titer viremia that WNV produces in horses (Bunning et al. 2002, Frost et al. 2012). Serological assays provide an alternative method of diagnosing infection with these viruses. However, cross-reactive antibodies induced to epitopes conserved between flaviviruses complicate the ability to accurately distinguish between infections of these closely related viruses (Hall et al. 1995, Knox et al. 2012).
The primary surface antigen on the flavivirus virion is the envelope (E) protein, which mediates cell attachment and entry, and is the major target of the humoral immune response to infection (Kimura and Ohyama 1988, Zhang et al. 2011). The protein structure of the E protein is divided into three distinct domains (DI, DII, and DIII) (Wu et al. 2003). DI and DII facilitate conformational changes during cellular entry and target membrane interaction during fusion (Allison et al. 2001, Modis et al. 2004), whereas DIII mediates interaction between viral receptor-binding domains and host cells receptors during attachment (van der Most et al. 1999, Kuhn et al. 2002, Mukhopadhyay et al. 2003, Chu et al. 2005). Flaviviral E-DIII can be expressed as a subunit recombinant protein that folds into an antigenically authentic and immunogenic structure (Martina et al. 2008).
Current diagnostic assays utilize the E protein as the target antigen; however, this protein contains many conserved epitopes, primarily located on domain II, and can elicit cross-reactive antibody responses. In contrast, DIII contains primarily type-specific epitopes (Sukupolvi-Petty et al. 2007). Indeed, a conformational epitope formed by E residues 306, 307, 330, and 332, located on the lateral surface of DIII in WNV, has been identified as a major inducer of virus-specific neutralizing antibodies (Sanchez et al. 2005, 2007).
Studies using WNV E-DIII have shown it to be a diagnostic protein of high specificity, and superior to whole-virus antigens (Beasley et al. 2004). In these studies, the DIII antigen allowed discrimination of specific antibody responses to WNV, and it allowed differentiation between WNV and yellow fever virus-positive sera (Holbrook et al. 2004, Batra et al. 2011). Ideally, a single diagnostic assay capable of quickly and accurately differentiating infections by closely related flaviviruses in regions of endemicity is desirable. As a step toward this goal, this study investigated the use of E-DIII as a diagnostic antigen to differentiate between MVEV and WNV infection in horses.
Materials and Methods
Expression and purification of recombinant E-DIII proteins
MVEV (AF161266.1) and WNV (AY274504.1) E-DIII proteins were expressed in BL21 (DE3) Escherichia coli (NEB) as a SUMO fusion protein using a Champion pET SUMO Expression System (ThermoFisher Scientific) as per the manufacturer's instructions. The expression of pET SUMO MVEV E-DIII and pET SUMO WNV E-DIII constructs was induced with 1.5 mM isopropyl-β-
Western blot
Cell pellets from both mock untransformed and transformed BL21 E. coli expressing MVEV or WNV rE-DIII protein were lysed in BS9 lysis buffer (120 mM NaCl, 50 mM H3BO3 + 1% Triton-X + 0.1% sodium dodecyl sulfate [SDS], pH 9.0) 4 h post-induction with IPTG. As a control to assess monoclonal antibody (mAb) binding, lysate from C6/36 (Aedes albopictus) cells infected with either MVEV or WNV were also included. C6/36 cell lysates were prepared similarly, using BS9 lysis buffer on uninfected cells or cells that had been infected with either virus for 5–7 days. Equal concentrations of protein with 4 × LDS Sample Buffer (Novex) were separated on 4–12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gels and transferred on to nitrocellulose membranes (GE Healthcare) before blocking with TENTC buffer (0.05 M Tris-HCl pH 8.0, 1 mM EDTA, 0.15 M NaCl, 0.05% [v/v] Tween 20, 0.2% [w/v] casein). The recombinant proteins were detected by using anti-WNV E-DIII (3.91D) (Adams et al. 1995) and anti-MVEV E-DIII (8E7) (Hall et al. 1990) specific mAbs. Bound antibody was detected by using horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG, and it was visualized by using DAB staining (0.6 mg/mL 3,3′-diaminobenzidine +0.06% [v/v] H2O2 in phosphate-buffered saline [PBS; pH 7.2]).
Coomassie blue staining
Proteins were separated on a 4–12% SDS-PAGE gel as earlier and immersed in Coomassie blue staining solution (3 × 10−3 M Coomassie Brilliant blue R250 [Bio-Rad], 45% methanol, 45% H2O, 10% glacial acetic acid) for 1 h. The gel was then rinsed several times in de-staining solution (45% methanol, 45% H2O, 10% glacial acetic acid) before visualization.
Horse serum samples
Equine serum samples were selected from a range of naturally or experimentally infected cohorts. Reference WNV-immune serum (animals 1–5) were obtained from equine infection studies conducted in the United States (Hobson-Peters et al. 2008) (Table 1). Horses experimentally infected with WNVNY99 were bled on the day of infection (Pre-) and ∼21–26 days postinfection (Post-). Serum samples (animals 13–43) were obtained from horses naturally infected with WNV in North and Central America. A panel of serum samples containing multiple bleeds from 34 horses naturally exposed during the WNV outbreak in south-eastern Australia in 2011 were also obtained for this study (Table 2). The WNV IgM titers for these samples were assessed by using a WNV IgM Capture enzyme-linked immunosorbent assay (ELISA) as per the manufacturer's instructions (IDEXX Laboratories, Westbrook, ME). WNV IgM ELISA values ≥3 were considered positive, between 2 and <3, inconclusive, and <2 negative. Previously characterized sera from WNV- or MVEV-immune or flavivirus-immune horses were obtained from animals naturally infected in south-eastern Queensland or the Northern Territory, Australia (Prow et al. 2013, Barton and Bielefeldt-Ohmann 2017). Sera from five horses characterized post-vaccination in a Japanese encephalitis virus (JEV) vaccine trial were also included in this study (Bielefeldt-Ohmann et al. 2014). Flavivirus negative sera comprised 10 samples of well-characterized sera from animals with no detectable antibody to any flavivirus.
A fourfold difference in neutralization titer was used to differentiate viral infection.
rE-DIII ELISA results are the mean of three replicates with an average BSA control subtracted to normalize background binding.
WNV horses previously characterized in Hobson-Peters et al. (2008).
Interpretation based on consensus of bELISA and VNT data for that sample.
MVEV control sample No. T143; previously characterized in Prow et al. (2013).
Average optical density of a representative sample of nine naive equine serum samples (Supplementary Table S1).
bELISA, blocking ELISA; BSA, bovine serum albumin; ELISA, enzyme-linked immunosorbent assay; Flavi, flavivirus positive; MVEV, Murray Valley encephalitis virus; N/A, not applicable; Neg, negative; rE-DIII, recombinant E-DIII; VNT, virus neutralization test; WNV, West Nile virus.
A positive threshold >4 and a fourfold difference in neutralization titer was used to differentiate viral infection. N/A for onset—exact date of onset is unavailable; however, the first sample was collected within 2–3 days of the first observable clinical signs of infection.
Interpretation based on consensus of WNV IgM, bELISA, and VNT data for that sample.
rE-DIII ELISA results are the mean of three replicates with an average BSA control subtracted to normalize background binding.
Enzyme-linked immunosorbent assay
To determine the optimal concentration of purified MVEV and WNV rE-DIII for coating plates in the ELISA, serial twofold dilutions were tested with anti-E-DIII mAbs and concentrations that produced an optical density (OD) of ∼1.0 were selected. A minimum dilution of 1/50 of horse serum was chosen for optimal sensitivity and minimal background reaction based on our previous published data (Hobson-Peters et al. 2011).
In the optimized ELISA format, rE-DIII (0.3 μg/mL for both antigens) in 50 μL coating buffer (50 mM NaHCO3, 50 mM Na2CO3, pH 9.6) was added to the wells of high-binding 96-well plates (Greiner) and incubated overnight at 4°C. The plates were blocked with TENTC buffer for 1 h at 28°C. Serum samples diluted 1/50 in 50 μL TENTC buffer were then applied and similarly incubated. The wells were washed with PBS-T (PBS, 0.05% [v/v] Tween 20), before applying 50 μL HRP-conjugated rabbit anti-horse (Sigma-Aldrich) IgG and incubated for 1 h at 28°C. After further washes with PBS-T, antibody binding was visualized by using 100 μL ABTS substrate [1 mM 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) [Sigma-Aldrich] + 3 mM H2O2 in 6 volumes of 0.1 M citric acid with 5 volumes of 0.2 M Na2HPO, pH 4.2] and incubating it for 1 h. The OD in each well was measured by using a plate reader at a wavelength of 405 nm. Bovine serum albumin (0.3 μg/mL) in coating buffer was used as the control antigen to account for nonspecific background readings, and it was subtracted from the sample OD readings. A threshold was set for samples specifically reacting with the WNV (OD 0.317) and MVEV (OD 0.285) rE-DIII antigens at 2 SD higher than the mean OD produced by a cohort of 10 flavivirus naive sera (Supplementary Table S1; Supplementary Data are available online at
Epitope-blocking ELISAs (bELISA) were performed by using methods previously described by Prow et al. (2013). Briefly, this assay involved coating U-bottomed 96-well polyvinyl chloride plates with an optimal concentration of flavivirus antigen diluted in 50 μL coating buffer and incubating overnight at 4°C. After the blocking and the addition of 50 μL serum samples (1/10), equal volumes of flavivirus pan-reactive (4G2) (Gentry et al. 1982, Broom et al. 1998), anti-MVEV (10C6) (Broom et al. 1998), or anti-WNV (3.1112G) (Blitvich et al. 2003) mAb were added to each well without removing the sera and further incubated for 1 h at 28°C. Antibody binding was visualized by using 50 μL HRP-conjugated goat anti-mouse IgG (Jackson ImmunoResearch) preabsorbed for minimal cross-reactivity to human, bovine, and horse serum proteins before visualization by using 100 μL ABTS substrate. Percent inhibitions were calculated at 100 − [OD (sample)/OD (negative control) × 100]. The cut-off for positivity was ≥30% inhibition.
Virus neutralization test
Virus neutralization tests (VNT) were performed in 96-well microtiter plates as previously described (Prow et al. 2013). Briefly, sera were titrated in doubling dilutions from 1/4 to 1/512 or 1/10 to 1/2560 in Dulbecco's modified Eagle's medium (DMEM) without fetal bovine serum (FBS) in a 96-well plate (Corning). Approximately 100 infectious units of MVEV (AF161266.1) or WNV (AY274504.1) diluted in DMEM with 2% FBS were added to each well containing the diluted serum and incubated at 37°C for 1 h. African green monkey kidney (Vero) cells resuspended in medium were then added to each well at a density of 2 × 105, and the plates were further incubated for 5 days at 37°C before examination for cytopathic effect (CPE). Cells were also added to either a 1/4 or a 1/10 dilution of each serum in the absence of virus to ensure that the samples were not toxic to the cells. The neutralization titer was expressed as the reciprocal of the highest serum dilution where CPE was recorded. Cells were fixed in the plates and subjected to an ELISA by using anti-MVEV (10C6) or anti-WNV (3.1112G) mAb, and a preadsorbed HRP-conjugated goat anti-mouse (Jackson ImmunoResearch) IgG secondary antibody.
Results
Successful expression and purification of bacterially expressed MVEV and WNV rE-DIII proteins
The MVEV and WNV pET SUMO plasmid constructs encoded residues 299 to 401 of the E protein from the corresponding virus (Fig. 1A). The SUMO fusion protein was used to enhance protein solubility and purification by using the incorporated N-terminal 6 × HIS tag. Assessment of the recombinant proteins as bacterial lysates using mAbs specific to MVEV or WNV E-DIII showed successful expression and antigenic authenticity of the respective rE-DIII proteins (Fig. 1B). Bioinformatic (Geneious) size prediction of the SUMO fusion is ∼13.4 kDa, whereas MVEV and WNV E-DIII are 10.9 and 11 kDa, respectively. Consistent with the predicted protein sizes, bands close to the expected size of ∼24 kDa were observed for both MVEV and WNV rE-DIII proteins. As a positive control, virus-infected C6/36 cell lysates were simultaneously probed with the mAbs and shown to detect the viral E protein at 50 kDa. Secondary bands ≥50 kDa were also observed in the bacterial lysates and may be indicative of dimer- and trimerization of the rE-DIII fusion proteins. No binding was observed in any of the mock control samples.
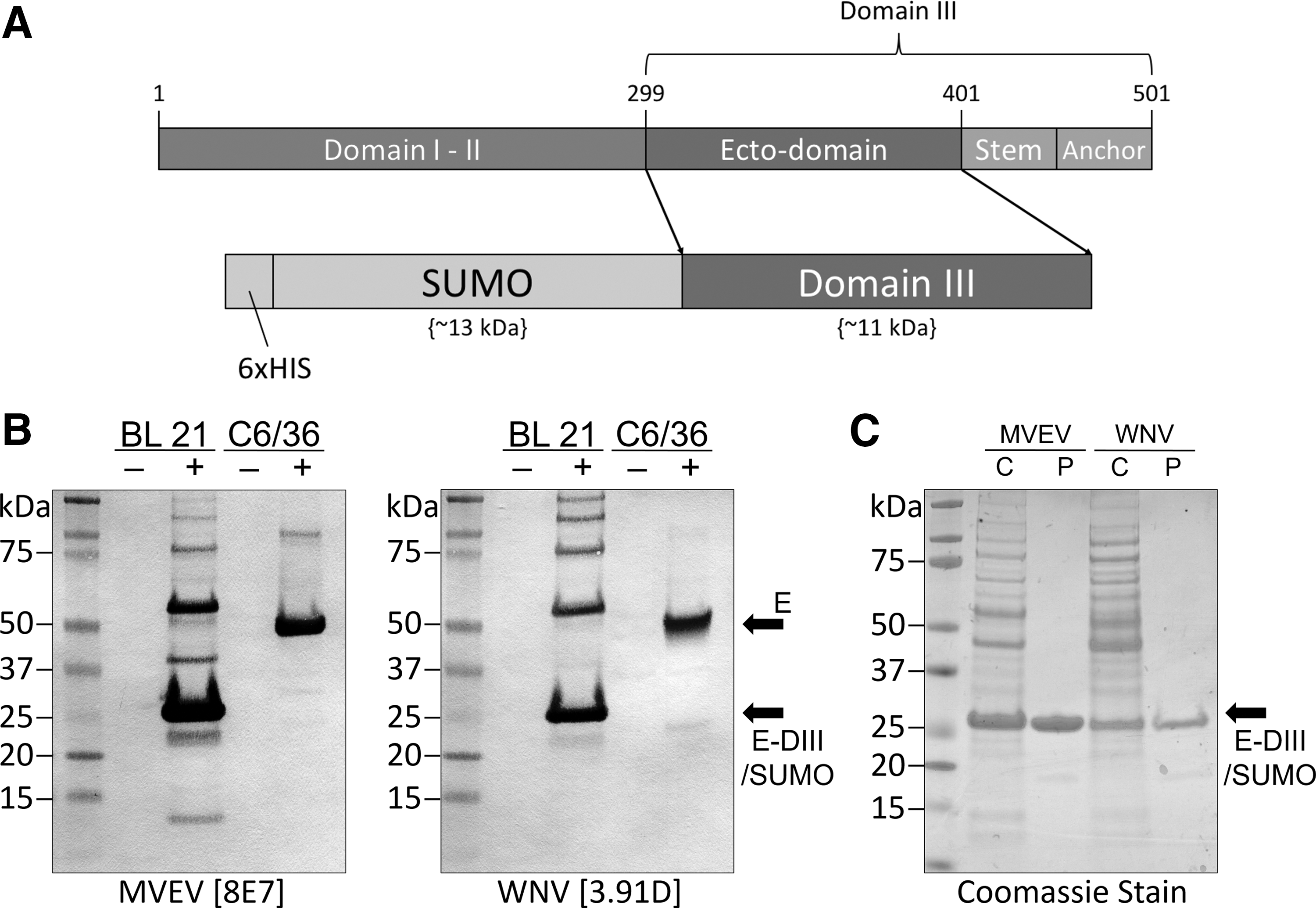
Expression and purification of rE-DIII protein of MVEV and WNV.
After purification of the rE-DIII proteins using IMAC and SEC, the purity of the proteins was analyzed on SDS-PAGE by Coomassie staining. Comparison of crude BL21 E. coli cell lysate to the final purified antigen showed a highly pure preparation for both MVEV and WNV rE-DIII proteins (Fig. 1C). Protein yields from a single 500 mL bacterial culture expressing either MVEV or WNV rE-DIII reached up to 0.3 mg/mL.
Reaction of sera from horses experimentally infected with WNV or naturally exposed to WNV during the NSW 2011 outbreak in the rE-DIII ELISA
To determine the efficacy of using rE-DIII proteins as antigens in ELISA to differentiate viral infection, we initially employed pre- and post-inoculation serum samples from horses experimentally infected with NY99 strain of WNV (Table 1). Results from the rE-DIII ELISA for all seven prechallenge sera available were consistent with the WNV neutralization assay and bELISAs. Of the eight post-WNV challenge sera, four reacted in the rE-DIII ELISA, with two (Post-5 and Post-32) giving an OD just below the threshold for a positive sample, but still showing a notably stronger reaction to WNV rE-DIII than to MVEV rE-DIII. Of these four positive reactors, the rE-DIII ELISA correctly differentiated three of these sera as being WNV positive, with the fourth sample yielding almost identical ODs for WNV and MVEV rE-DIII. In comparison, there was 100% agreement between the WNV bELISA and WNV virus neutralization assay. A single reference sample from an MVEV-immune horse, previously characterized by Prow et al. (2013), was specific for MVEV in each test. In summary, the WNV rE-DIII ELISA had a sensitivity of 57% when compared with bELISA and VNT for the cohort of WNV-challenged horses. When the data of both rE-DIII ELISAs were taken into consideration, three out of the four positive reactors were correctly identified as WNV positive.
To further determine the sensitivity and specificity of the rE-DIII antigens, multiple serum samples from 34 horses naturally exposed during the 2011 WNV outbreak in south-eastern Australia (2–4 samples from each animal taken over a 1–5 week period after presentation with clinical signs) were also tested against the WNV and MVEV rE-DIII antigens in ELISA in parallel with VNT, IgM ELISA, and bELISAs (Table 2). Of the 34 horses, 27 (79%) showed significant WNV IgM titers on 1 or more samples, whereas serum reactivity from 24 of the 34 animals (71%) to WNV E-DIII suggested a recent or ongoing WNV infection by at least the final bleed. Of the remaining 10 animals, 2 were suggestive of MVEV infection (samples 26, 27), 4 were equivocal for both viruses (samples 11, 22, 28, 34), and 4 were negative to both WNV and MVEV rE-DIII antigens in all bleeds (samples 10, 16 18, 33). Similarly, 18 of the animals (53%) were clearly positive for WNV by neutralization assay (≥4-fold rise in WNV titer and no significant change in MVEV titer), 13 were equivocal for both WNV and MVEV, and 4 were negative or trace reactors only (titer ≤4) for both viruses by the final bleed. In comparison, all animals were positive for flavivirus reactive antibodies in the flavivirus pan-reactive bELISA, with serum from 18 animals (56%) only reactive (percent inhibition >30%) in the WNV-specific bELISA by the final bleed. Of the remaining 16 horses, 15 were equivocal for WNV or MVEV infection by the WNV- and MVEV-specific bELISAs by the final bleed, and a single animal (sample 33) was negative for antibody in both MVEV and WNV bELISAs for both samples taken.
IgM titers were generally observed to decrease over time after a peak close to the initial sample collection date. Although less sensitive than the IgM ELISA, the rE-DIII ELISA results compared well with the VNT and the bELISA for specificity. The rE-DIII ELISA also identified samples as possible MVEV positives (e.g., samples 26–27), which was supported by the bELISA data, but not by the VNT results. Of interest, animal 33 was negative in both MVEV and WNV bELISAs and in both rE-DIII assays but was clearly positive for neutralizing antibodies to WNV in serum from the final bleed.
The results cited earlier indicated that the rE-DIII ELISA showed an overall sensitivity of 88% for the horses naturally exposed to the NSW WNV outbreak in 2011 when compared with the consensus of the VNT and bELISA.
Reaction of sera from horses naturally exposed to WNV, MVEV, and other heterologous flaviviruses in the rE-DIII ELISA
An additional 16 horses from the Northern Territory and south-east Queensland previously characterized by Prow et al. (2013), Barton and Bielefeldt-Ohmann (2017), and Hobson-Peters et al. (2008) were also surveyed (Table 3). Of sera from these animals, 15 (93%) showed neutralizing antibody to either MVEV or WNV or both, with sera from 4 (27%) showing a significantly higher titer (≥4-fold titer) to MVEV, and 4 (27%) neutralizing WNV more strongly. Titers of the remaining seven were equivocal for the two viruses. By comparison, all sera tested positive for flavivirus-reactive antibodies in the flavivirus pan-reactive bELISA, of which 10 (63%) were deemed positive to WNV and 6 (38%) to MVEV in the virus-specific bELISAs. All eight sera deemed positive to either MVEV or WNV by VNT showed the same result in the rE-DIII ELISAs. Of the four sera that gave a very low titer by VNT (≤20), three were negative and one was positive for MVEV (T141) by rE-DIII ELISA. By comparison, of the six sera positive to either MVEV or WNV by bELISA, five gave the same result in the rE-DIII ELISAs. Overall, the rE-DIII ELISA was able to consistently reflect the diagnostic consensus reached by using the bELISA and VNT.
A fourfold difference in neutralization titer was used to differentiate viral infection.
Interpretation based on consensus of bELISA and VNT data for that sample.
rE-DIII ELISA results are the mean of three replicates with an average BSA control subtracted to normalize background binding.
Horses previously tested in Barton and Bielefeldt-Ohmann (2017) also contained neutralizing titers to Ross River virus.
Horses previously tested in Prow et al. (2013).
Serum samples also contained neutralizing titers <80 to JEV.
JEV, Japanese encephalitis virus.
To further determine the impact of cross-reactive flavivirus-immune sera on MVEV and WNV rE-DIII antigens, a panel of sera from horses vaccinated with an inactivated JEV vaccine (Bielefeldt-Ohmann et al. 2014) were also tested in the ELISA (Table 4). Of the five sera obtained from JEV-vaccinated horses, all yielded moderate titers to JEV in VNT and also tested positive to both rE-DIII antigens in ELISA. Three samples (14F11, 15F11, and 17F11) reacted to significantly higher levels to MVEV rE-DIII, consistent with previous reports that equine JEV vaccination induces cross-reactive responses to both MVEV and WNV, with responses being generally higher to the former (Bielefeldt-Ohmann et al. 2014). Of interest, no neutralizing antibody to MVEV or WNV was detected in these sera.
A fourfold difference in neutralization titer was used to differentiate viral infection.
Interpretation based on consensus of bELISA and VNT data for that sample.
rE-DIII ELISA results are the mean of three replicates with an average BSA control subtracted to normalize background binding.
Horses previously tested in Bielefeldt-Ohmann et al. (2014).
Horses previously tested in Prow et al. (2013).
?, No clear interpretation; KOKV, Kokobera virus; NT, not tested; STRV, Stratford virus.
Ten horse sera with moderate neutralizing titers to the flaviviruses Stratford virus (STRV) or Kokobera virus (KOKV) (Prow et al. 2013) were similarly tested on both rE-DIII antigens in ELISA. Although none of the sera exhibited neutralizing antibodies specific to MVEV or WNV, three showed reactivity to either MVEV or WNV rE-DIII, with two showing higher levels to MVEV and one reacting equally to both. One sample, which also weakly neutralized WNV (T112), had greater binding to WNV in the rE-DIII ELISA, but it did not meet the positive threshold. The results for T114 and T118 were consistent with that of the bELISA data.
The sensitivity of the rE-DIII ELISA was consistent with bELISA and VNT results of naturally exposed horses. However, a reduced specificity was observed primarily due to the JEV-vaccinated horses, suggesting that a cross-reaction may occur in animals infected with or vaccinated against heterologous flaviviruses.
Discussion
Flavivirus infections are capable of generating cross-reactive antibodies to conserved antigenic determinants in the E protein, especially in E-DII (Crill and Chang 2004, Lai et al. 2008). This can compromise serodiagnostic assays attempting to differentiate infection between closely related flaviviruses. Therefore, improvements in the specificity of these diagnostic assays are required. This study assessed the utility of rE-DIII as a target antigen to serologically differentiate between infections of horses with either MVEV or WNV. E-DIII has previously been reported to be highly antigenic, and it predominantly comprised subcomplex and type-specific epitopes that induce neutralizing antibodies (Rey et al. 1995, Beasley and Barrett 2002, Sukupolvi-Petty et al. 2007). Our findings corroborate previous studies, which demonstrated the usefulness of the E-DIII antigen for differentiating between antibody responses to related flaviviruses such as WNV, Wesselsbron virus, and tick-borne encephalitis (Beasley et al. 2004, Holbrook et al. 2004, Mathengtheng and Burt 2014).
Based on the diagnostic consensus of the bELISA and VNT, the rE-DIII assay showed a reduced sensitivity for diagnosing WNV infections in horses experimentally infected via the intradermal route with the North American WNV strain NY99. In animals naturally exposed to the WNV strain associated with the 2011 outbreak of equine encephalitis, the rE-DIII ELISA displayed specificity and sensitivity similar to both VNT and bELISA. The WNV-specific bELISA has previously been shown to be highly effective for detecting WNV-specific antibody responses in horses, which target the NS1 protein (Broom et al. 1998, Blitvich et al. 2003, Ledermann et al. 2011, Prow et al. 2013). It is also worth noting that some samples with no detectable neutralizing antibody to either MVEV or WNV by VNT produced reaction patterns to the rE-DIII antigens that corresponded to reactions in the virus-specific bELISAs, suggesting that a lack of correlation with VNT results is not necessarily indicative of a lack of specificity. An inverse correlation of IgM titers to rE-DIII readings was observed after the onset of illness in serum from horses naturally exposed during the 2011 WNV outbreak. This suggests that using rE-DIII with an IgG secondary antibody is effective at detecting the IgG isotype switch in horse serum at a relatively early stage postinfection. The rE-DIII antigen should also be investigated as a possible antigen for use in a WNV-specific IgM assay, particularly as the WNV IgM assay has been shown to be highly sensitive and specific in detecting early infection (Long et al. 2006, Ledermann et al. 2011).
Although the use of rE-DIII antigens in ELISA to exclude serum from horses naturally exposed to WNV and MVEV was consistent with results from the bELISA and VNT, horses immune to heterologous flaviviruses provided mixed results. Three out of the 10 sera with moderate titers to the Kokobera group viruses, but negative to MVEV and WNV by VNT, showed reactivity to either MVEV only or both MVEV and WNV rE-DIII antigens at the dilution tested. However, two out of these rE-DIII reactors produced corresponding results in the bELISA. Since KOKV and STRV are enzootic to most regions of Australia and commonly infect horses, this is an important issue to consider for flavivirus diagnosis in these animals (Prow et al. 2013). However, although KOKV infection can produce a polyarticular disease in humans, there have been no reports of its association with disease in horses (Prow et al. 2013). Although KOKV and STRV were originally included in the JEV serological group, subsequent phenotypic and genetic characterization (Hall et al. 1991, Poidinger et al. 1996, Kuno et al. 1998) led to their reclassification into a separate virus group within the flavivirus genus (Van Regenmortel 2000).
Further, all sera from horses vaccinated against JEV cross-reacted to MVEV and WNV rE-DIII antigens in ELISA, consistent with our previous studies showing cross-neutralizing responses to MVEV and WNV, generally stronger to the former (Bielefeldt-Ohmann et al. 2014). Interestingly, no similar cross-reactive responses were detected in these sera in this study. The lack of cross-reactivity by the sera from JEV-vaccinated animals in the MVEV- and WNV-specific bELISAs is also consistent with the use of the NS1 antigen in these assays, which is not a component of the purified and inactivated JEV vaccine.
Despite the limitations identified in this study, the capacity for large-scale expression of rE-DIII in bacteria, and its inherent stability makes it a useful candidate for the development of rapid diagnostic formats (Bhardwaj et al. 2001, White et al. 2003, Holbrook et al. 2004). The rE-DIII antigen may also be suitable for multiplex assays such as multiplex immune assays (e.g., Luminex), which can provide a rapid, inexpensive, and accurate approach for screening equine sera (Balasuriya et al. 2006, Hobson-Peters 2012). Finally, the rE-DIII-based ELISA described here will also be particularly useful for regional labs with limited resources. The antigens can be safely shipped, require no special biocontainment, and are stable at −20°C.
Conclusion
Cross-reactive antibodies remain challenging and complicate the ability to distinguish between infections of MVEV and WNV in horses. This study has shown the utility of rE-DIII as an antigen allowing differentiation between clinically relevant flavivirus infections in Australia. Further evaluation of these antigens, possibly including other flaviviruses such as JEV, on a larger collection of serum samples is necessary. These findings also indicate that future studies that include rE-DIII antigens in multiplex diagnostic assays or other formats for flavivirus infections are warranted.
Footnotes
Acknowledgments
The authors would like to thank Dr. Daniel Watterson and Dr. Keith Chappell (University of Queensland) for their expert technical advice and assistance. The technical assistance of the staff of the Virology Laboratory at Elizabeth Macarthur Agricultural Institute in undertaking testing of the NSW field sera is also greatly appreciated. This study was funded by the Australian Research Council (ARC LP120100686) and an Australian Government Research Training Program Scholarship to T.B.H.P.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.