
Abstract
There are two distinct lineages of ticks, Rhipicephalus sanguineus, in South America: tropical and temperate lineages. Only the tropical lineage is recognized as competent vector for Ehrlichia canis. The epidemiological data of canine monocytic ehrlichiosis is congruent with the distribution of the two lineages of R. sanguineus. Herein, we report the infection of R. sanguineus (tropical lineage) cell cultures with E. canis, after cryopreservation. R. sanguineus (tropical lineage) cell identity was confirmed by sequencing using a 16S rDNA gene fragment. Tick cell cultures were prepared in L-15B medium supplemented with 10%, 15%, and 20% Fetal Bovine Serum (FBS), and 10% of Tryptose Phosphate Broth (TPB). Cell cultures developed better at the concentration of 20% of FBS. Cultures in the fifth harvest (approximately 7 months later) were selected for the first infections. Optimal R. sanguineus cell growth and adhesion was observed (5.0 × 106 cells/mL, and the population doubling time every 57 h). Once infected with E. canis, the cultures were maintained in L-15B medium supplemented with 2% and 5% of FBS fortified with iron and 10% TPB. Infected cells were also cryopreserved. DNA was extracted from infected and noninfected cells and analyzed using quantitative real-time PCR targeting the E. canis-dsb gene. Primary culture of the fifth passage was infected by E. canis and it maintained the pathogen for at least 40 days before partial cell destruction. Subcultures of infected cells (fresh and cryopreserved cultures) onto new tick cell cultures were successful. The E. canis infection was confirmed by real-time PCR and light and transmission electron microscopy. The R. sanguineus (tropical lineage) cells infected with E. canis successfully infected new tick cell cultures, showing that these cells could be an alternative substrate for maintenance of this pathogen.
Introduction
T
Rhipicephalus sanguineus (s.l.) ticks are considered the main vector of Ehrlichia canis (Anaplasmataceae) that is responsible by canine monocytic ehrlichiosis (CME) (Lewis et al. 1977). This disease has been described around the world in tropical and subtropical regions (Vieira et al. 2011), but, CME appears to be particularly prevalent in tropical regions where it is principally vectored by R. sanguineus (s.l.) ticks (Cicuttin et al. 2015). The occurrence of CME in tropical regions of the South America (Moraes-Filho et al. 2015), is related to the difference in the vector competence between populations of R. sanguineus (s.l.). In this context, populations of ticks belonging to the tropical lineage are highly competent vectors of E. canis. On the other hand, ticks belonging to the temperate lineage, distributed in South America below an ecotonal zone situated between 24° and 25° of south latitude (Nava et al. 2012), are incompetent vectors of E. canis, coinciding with scarcity or absence of CME in that area.
Ehrlichia canis is a pleomorphic Gram-negative, obligatory, intracellular bacterium found isolated (elementary bodies) or in compact inclusions within the cytoplasmic vacuoles (morulae) of monocytes and macrophages (Dumler et al. 2001). Monocytes and neutrophils kill invading microorganisms by oxygen-independent mechanisms, such as fusion of the phagosomes containing bacteria with granules containing both antimicrobial peptides such as defensins or lysozymes and lysosomal hydrolytic enzymes or through sequestering vital nutrients such as iron (Rikihisa 2010).
Some of the pathogens transmitted by ticks are very difficult to grow in traditional culture media mainly due to their requirements, and, according to Stewart (2012), the bacteria that can be grown in the laboratory are only a small fraction of the total diversity that exists in nature. E. canis is one of them. The proliferation and propagation process of Ehrlichia sp. in host cells depends on the nutrients' acquisition and lysosomal evasion, involving activation of sequential signaling events modifying the host proteins required for their entry and their establishment (Rikihisa 2006).
The cultivation of E. canis in monocyte cell cultures derived from dogs in the acute phase of the disease was reported since 70's, but they were short-term cultivation (Nyindo et al. 1971). Such cultures were limited due to maintenance of the organism by sequential transfer of infected monocytes and the continuous supply of cells was also limited because they were primary cultures. Nevertheless, the laboratory diagnosis improved immensely after propagating E. canis serially from primary cultures of monocytes, mainly the indirect fluorescent antibody test (Iqbal et al. 1994).
In 1975 the first continuous tick cell culture of Rhipicephalus appendiculatus Neumann 1901 (Varma et al. 1975) was established and since then, ticks cell cultures have been successfully used in isolation (Munderloh et al. 2003, Cabezas-Cruz et al. 2016), and propagation of several pathogens, including virus (Lawrie et al. 2004, Mansfield et al. 2017) and bacteria (Munderloh et al. 1996a, Policastro et al. 1997, Alberdi et al. 2016). The use of “in vitro” culture systems has shown applicability for several studies, such as morphological, genetic, proteomic, transcriptional genomic, and protein expression, allowing comparisons between host cells and vectors (Bell-Sakyi et al. 2007).
The first culture and propagation of E. canis “in vitro” was carried out on cells originating from canine monocytes, known as DH82 (Dawson et al. 1991). Later, Ewing et al. (1995) inoculated leukocytes infected with E. canis in cell culture of various tick species, including R. sanguineus s.l., but success was achieved in isolation only in the tick cell line of the nonvector of E. canis, Ixodes scapularis IDE8 (Munderloh et al. 1994). Afterward, a North American isolate and a South African isolate of E. canis were grown in another lineage of I. scapularis, ISE6 (Singu et al. 2006) and Ixodes ricinus Linnaeus, 1758 (Bell-Sakyi et al. 2007), respectively. Furthermore, Zweygarth et al. (2014) reported the isolation and propagation of two isolates of E. canis from South Africa and one from Spain on IDE8 cells from I. scapularis. However, only 2 years ago, for the first time, the infection of E. canis in tick cell culture of R. sanguineus s.l (RSE8) (Ferrolho et al. 2016) was observed. Although, the RSE8 tick cell lines were established in laboratories in the USA, their origin is unknown (Kurtti et al. 1982, Yunker et al. 1984). But, according to Ferrolho et al. (2016) the E. canis infection in RSE8 was maintained only for 4 weeks and attempts to subculture the bacteria onto fresh RSE8 cultures were unsuccessful, and a second R. sanguineus s.l. cell line, RML-RSE, proved completely refractory to the infection. On the other hand E. canis has successfully infected other tick cell lines (nonvector) (Zweygarth et al. 2014, Bell-Sakyi et al. 2018).
Then, the aim of this study was to prepare in vitro primary cultures from embryonic eggs of R. sanguineus s.l. tropical lineage (tick-vector) and test their use as a substrate for maintenance of E. canis (Jaboticabal strain). The E. canis cultivation in its vector embryonic cells, R. sanguineus (tropical lineage), even if it is in primary cultures, may open up new research opportunities to increase the knowledge of the E. canis life cycle in the tick. In addition, this knowledge could help to understand the interaction between the bacteria and the tick at the cellular and molecular level, leading to new strategies for the control and prevention of EMC.
Materials and Methods
Origin of ticks and ethics approval
Ticks were collected from dogs from Uberlândia city, state of Minas Gerais, Brazil (18°55’07”S 48°16’38”W). The colonies have been maintained in the laboratory of Ixodology, Federal University of Uberlândia since 2005. From time to time, some new specimens from the same locality were introduced into the colony to avoid effects of inbreeding. The maintenance of the colonies was authorized by the Ethics Committee of that Institution (registration CEUA/UFU 033/14). Engorged females were recorded in the reception of Animals of the Butantan Institute, receiving the tracking number 2014067798, and after they laid eggs, they were deposited into the Acari Collection at the same Institution under the number IBSP 11.734. The tick cell cultures were authorized by the Conselho de Gestão do Patrimônio Genético (CGEN No. 010214/2015-1).
Preparation of primary tick cell cultures
The engorged females were washed with alcohol 70% and immersed in a 1% benzalkonium chloride solution (Polyorganic, São Paulo, SP, Brazil) for 15 min. Then, the specimens were rinsed in sterile distilled water (50 mL H2O containing 100 μL of Penicillin/Streptomycin and 50 μL Amphotericin B) (Vitrocell Embriolife, Campinas, SP, Brazil) for 5 min, following previous protocols (Pudney et al. 1973, Bhat and Yunker 1977, Kurtti et al. 1983). After drying in sterile gauze, the females were individually placed into sterile Petri dishes and kept in a biological oxygen demand incubator at temperature of 27°C ± 1°C, 80–85% humidity in darkness to perform ovipositions. The eggs were daily collected, and when they reached the age (14–15 days of age) they were transferred to a small Petri dish and gently crushed in complete L-15B medium (Vitrocell Embriolife), as described in a previous protocol (Munderloh and Kurtti, 1989). Tissues and egg shells were filtered in sterile gauze, resuspended in L-15B medium, and centrifuged at 100 g for 8 min. The pellet was resuspended in 4 mL of complete L-15B medium with concentrations of 10%, 15%, and 20% of Fetal Bovine Serum (FBS; Vitrocell Embriolife) and 10% of Tryptose Phosphate Broth (Vitrocell Embriolife), according to Kurtti et al. (1982). The cell suspension was added with 4 μL of Penicillin/Streptomycin (Vitrocell Embriolife) and 4 μL of Amphotericin B (Vitrocell Embriolife).
The cultures were incubated at 30°C and the medium was replaced weekly, without antibiotics after the first week. The cellular adhesion under each concentration of FBS (Vitrocell Embriolife) was monitored using an inverted light microscope (Nikon, Eclipse TS100). When confluence was reached 90–100% (∼2 months later), the cells were removed from the flask by scraping slightly. After centrifugation in a Falcon tube with 6 mL of complete L-15B medium (for 8 min at 100 g), the supernatant was discarded, the pellet was resuspended in 8 mL of L-15B medium, and divided into two new flasks. Cultures in the fifth harvest (∼7 months later) were selected for the first infections.
Tick cell identity
Cell samples (4 mL) of R. sanguineus (tropical lineage) were scraped from the flasks (25 cm2) and processed to DNA extraction using the DNeasy Tissue Extraction Kit (Qiagen®, Chatsworth, CA), following the manufacturer's instructions. The extracted samples were submitted to conventional PCR based on mitochondrial 16S rRNA (Black and Piesman 1994), for confirming the cell identity. The mixture contained PCR buffer 10 × (Life Technologies®, Carlsbad, CA), MgCl2 1.0 mM (Life Technologies), deoxynucleotide triphosphate 0.2 mM (dNTPs) (Life Technologies), 1.5 U Taq DNA Polymerase (Life Technologies), and 0.5 of primers 16S + 1 (5′-CTG CTC AAT GAT TTT TTA AAT TGC TGT GG-3′) e 16S - 1 (5′-CCG GTC TGA ACT CAG ATC AAG T- 3′) (Integrated DNA Technologies®, Coralville, IA), to final volume 25 μL. Amplified products were purified using the Silica Bead DNA Gel Extraction Kit (Thermo Fisher Scientific™, Waltham, MA) and sequenced using the BigDye™ Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific) and the ABI PRISM 310 DNA Analyzer (Applied Biosystems™, Foster City, CA) (Sanger et al. 1977). The primers were the same as those used for PCR. The cell identity was confirmed using the Blast program (
Infection of tick cell cultures by E. canis extracted from DH82 cells
First, DH82 cells (canine macrophage cell line) infected with E. canis (strain Jaboticabal–GenBank accession number DQ460716) (Aguiar et al. 2008) were centrifuged (500 g for 10 min) and 1 mL of the supernatant rich with bacteria (quantified by qPCR described afterward) was transferred to a Falcon tube containing L-15B medium without FBS and antibiotics. The Falcon tube was inverted a few times for homogenization and centrifuged at room temperature for 15 min (at 400 g). The supernatant was discarded and 2 mL of L-15B medium with 10% TPB without FBS and antibiotics was added. The suspension was again homogenized and centrifuged as above. This procedure was repeated three times. After the second centrifugation, 100 μL was removed to prepare slides that were submitted to the cytocentrifugation (Cytospin 4; Thermo Scientific, Waltham, MA) at 100 g for 3 min. The slides were stained using the Quick Panoptic Kit (LaborClin®Pinhais, PR, Brazil).
Ehrlichia canis extracted from DH82 cells, after the third centrifugation, was inoculated in two flasks of 25 cm2 with cells of R. sanguineus (tropical lineage) (fifth subculture) in L-15B medium supplemented with 2% and 5% of FBS fortified with iron (trademark Hyclone®; Thermo Scientific; catalog number SH30072.04, lot AYA56025). Both concentrations (2% and 5%) of FBS fortified with iron were tested to establish an optimal concentration to be employed. The flasks were maintained at 30°C. The L-15B medium supplemented with 2% and 5% of FBS fortified with iron (trademark Hyclone; Thermo Scientific; catalog number SH30072.04, lot AYA56025) was partially replaced weekly.
Cryopreservation of semipurified bacteria and propagation in new tick cells
After 4 weeks, R. sanguineus (tropical lineage)-positive cell cultures, with E. canis, were propagated in fresh tick cell cultures or they were ultracentrifuged at 9000 g to extract the semipurified pathogens before cryopreservation. The tick cells infected with E. canis were removed from the flask by scraping and centrifuged at 4000 g for 15 min. The supernatant was again centrifuged at 9000 g for 15 min and the pellet was resuspended in 1 mL of L-15B medium supplemented with 25% FBS (Vitrocell Embriolife) and 10% DMSO. The suspension was placed in cryovials that were kept in a Nalgene™ Cryo 1°C freezing container (Thermo Fisher Scientific, Cat. No. 5100-001) to achieve a −1°C/min rate cooling, and then frozen in liquid nitrogen at −196°C.
For defrosting, the cryotubes with semipurified E. canis were thawed quickly and placed in 5 mL FBS (Vitrocell Embriolife). The cryotubes were then centrifuged for 15 min at 9000 g. The supernatant was discarded and the pellet was resuspended in 2 mL L-15B medium with 2% and 5% of FBS fortified with iron (Hyclone; Thermo Scientific; catalog number SH30072.04, lot AYA56025) and distributed onto new fresh tick cells.
Cell growth curves
The cell-uninfected R. sanguineus (tropical lineage) cells with 20% of FBS (Vitrocell Embriolife), and tick cells uninfected (controls) and infected with E. canis with 5% and 2% of FBS fortified with iron (Hyclone; Thermo Scientific; catalog number SH30072.04, lot AYA56025), were scraped from the flasks (25 cm2). They were counted in digital automatic cell counter (TC 10™; Bio-Rad) with Trypan Blue dye exclusion and plated in a six-well plate to determine the cell growth every 24 h for 10 days. It was incubated at 30°C. The maximum specific growth rate (μmax) was determined by a nonlinear regression curve analysis and the population doubling time was also evaluated (Munderloh and Kurtti 1989).
Quantitative real-time PCR
Before the infection, a previously described quantitative real-time PCR (qPCR) protocol was performed to E. canis based on dsb gene (Doyle et al. 2005), to rule out the presence of the bacteria in the cell cultures of R. sanguineus (tropical lineage). The qPCR was also performed to DH82 cells infected with E. canis to quantify the pathogen per μL, following MIQE guidelines (Bustin et al. 2009).
Sample of DH82 cells infected with E. canis (1 mL) and sample cells (4 mL) of uninfected ticks (control) and infected with E. canis were scraped from the flasks (25 cm2) and processed individually to DNA extraction using the DNeasy Tissue Extraction Kit (Qiagen), following the manufacturer's instructions.
Transmission electron microscopy
Samples of the tick cells that were uninfected (control) and E. canis-infected tick cells were fixed in Karnovsky solution in 0.1 M cacodylate buffer at pH 7.2 for 2 h. Then, cells were postfixed in osmium tetroxide solution 1% in the same buffer for 1 h. After washing, the samples were immersed in 0.5% uranyl acetate solution with 13.3% sucrose, and stored at 4°C for 24 h. Dehydration was performed using an ethanol series (50%, 70%, 80%, 90%, 95%, and twice 100%). After the dehydration, the samples were infiltrated with 1:1 mixture of propylene oxide with Epon resin for 2–3 h. The cell suspensions were transferred to a BEEM capsule with pure resin and centrifuged at 900 g for 5 min. After replacement of the resin and centrifugation, the capsules containing pellet of cells, were dried at 60°C for polymerization for 2–3 days. Ultrathin sections (60–70 nm) were placed on copper grids, contrasted with aqueous solution of uranyl acetate 2% for 10 min, and then with lead citrate for 3–5 min (Reynolds 1963). Finally, it was examined under a transmission electron microscope (TEM) LEO 906E (Zeiss, Germany). The images were captured with a CCD camera Mega View III using the program iTEM (Universal TEM Imaging Platform; Olympus Soft Imaging Solutions GmbH, Germany), in the Laboratory of Electron Microscopy, Institute of Biosciences, Universidade Estadual Paulista Julio de Mesquita Filho (UNESP)–campus Rio Claro, state of São Paulo.
Results
Fifty engorged females were used resulting in 50 egg masses. The average weight of females and egg mass were 126 and 70 mg, respectively. Ten cell flasks of primary cultures of R. sanguineus were initially obtained, but three of them did not go forward, probably because the FBS concentrations used were of 10% (two flasks) and 15% (one flask), not allowing cell growth. The first subculture was carried out 60 days after the primary cell cultures when the number of total cells was 5.0 × 106 cells/mL (live 95%). This total number of cells/mL was slightly increased (6.0 × 106 cells/mL) and it was maintained in each passage (immediately before to split them). For this tick species, the growth and adhesion of the cells was observed only when the concentration of FBS was 20%.
Although, FBS concentration (20%) was good enough for R. sanguineus’ (tropical lineage) cell growth, the population doubling time every 57 h (Table 1) was not good for the keeping of infected cells with E. canis. When the primary tick cell cultures were inoculated with E. canis and maintained in L-15B medium with high FBS concentration (20%), the tick cells were not infected.
Cell Growth Parameters of Rhipicephalus sanguineus (Tropical Lineage) Uninfected and Infected with Ehrlichia canis, Under Different Fetal Bovine Serum Concentrations
Td (h−1), population doubling time (h); X max, maximal cell density (106 cell/mL−1); Tmax (days), time to reach maximal cell number.
Before E. canis inoculation in tick cell cultures, the quantification of the pathogen in 1 mL of DH82 cells by qPCR showed (mean Cq of 7.33 × 105 copies/μL. E. canis dsb gene copies/μL) (E = 98.3; r 2 = 0.931; slope = −3.536; Y intercept = 42.045).
The tick cells only got infected with E. canis after they had been kept in L-15B with 2% and 5% FBS fortified with iron (brand Hyclone). At these FBS fortified with iron concentrations, tick cell growth slowed (Fig. 1), offsetting the pathogen increase, and new cell invasion. Two tick cell cultures were kept without infection (control) with both concentrations (2% and 5%) of FBS fortified with iron (Table 1 and Fig. 1). The control cells became very vesiculated and entered into senescence after 1 month and a half.
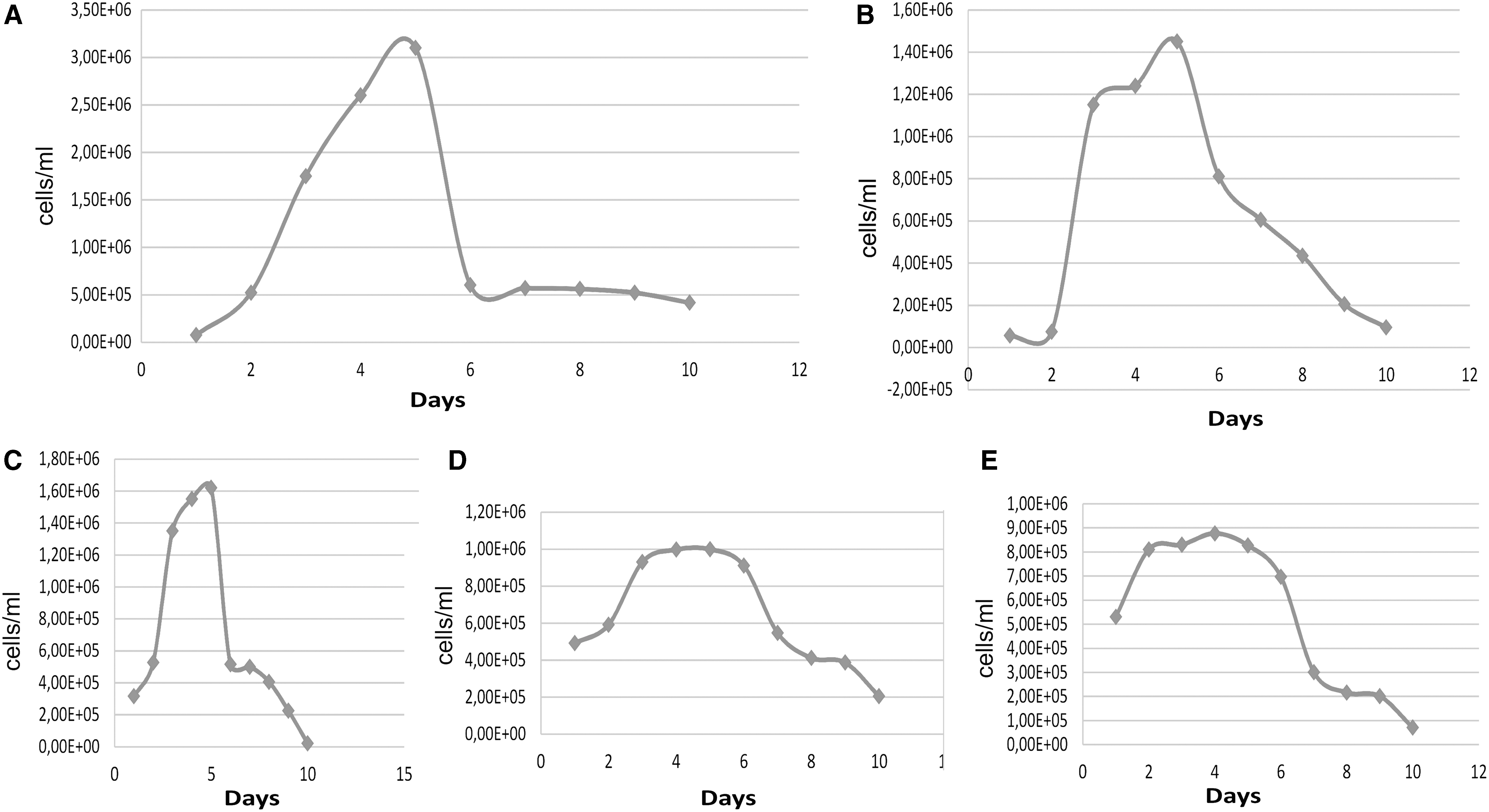
Rhipicephalus sanguineus (tropical lineage) cell growth.
The number of cells of R. sanguineus (tropical lineage), after 20 days of infection with E. canis, was 4.5 × 105 (live 66%) (15% infected cells in 200 counted cells). After 30 days, the number was 3.74 × 105 (live 60%) (30% infected cells in 200 counted cells). When the percentage of infected cells reached 30% (in 200 counted cells) in 30 days, an aliquot of 500 μL was propagated in uninfected R. sanguineus cell culture.
Subculture of infected cells onto new cell cultures was successful by transferring one-third of infected cultures into uninfected ones, as well as the cryopreserved infected cells, when propagated in fresh tick cell cultures (Figs. 2 and 3).


Transmission electron microscopy of cells from the fifth harvest of R. sanguineus (tropical lineage).
In this time, the identity of R. sanguineus cells was confirmed by sequencing a 460 pb fragment of 16S rRNA gene followed by BLAST analysis. The sequence obtained from R. sanguineus cells showed (Query cover 99%) identity with R. sanguineus (tropical lineage) from Uberlandia Municipality (GU553074). The sequence was deposited under the GenBank accession number MF187515.
Infection with E. canis was confirmed by qPCR in tick cell cultures obtained after propagation (mean Cq of 2.7 × 101 E. canis dsb gene copies/μL) and thawing followed by propagation (mean Cq of 2.43 × 101 E. canis dsb gene copies/μL) (E = 94.1; r 2 = 0.996; slope = −3.473; Y intercept = 41.720).
The infected tick cell cultures were propagated in continuous cultivation in vitro in five consecutive propagations after thawing.
The primary cultures of the fifth harvest were able to maintain the pathogen for at least 40 days before partial cell destruction. DH82 cells with E. canis and cell culture of R. sanguineus (tropical lineage) infected with E. canis are shown in Figure 2A. Morulae appear in cells from 15 days and 30 days postinfection (Fig. 2B, C) and also 30 days postinoculation after thawing (Fig. 2D). The TEM showed cells infected with E. canis (Fig. 3). The presence of E. canis morulae could be seen in the cell cultures of R. sanguineus (tropical lineage) 2–4 weeks postinfection after cryopreserving pathogens.
Discussion
In the present study, the primary cell cultures of R. sanguineus (tropical lineage) prepared in L-15B complete medium with FBS at 20% concentration showed cell growth and cell adhesion. A previous study showed that concentrations of 20% FBS and 10% TPB were more suitable for cellular adhesion and growth in R. sanguineus s.l. cell line RSE8 (Ferrolho et al. 2016), in agreement with the results observed in the present study with cell cultures of R. sanguineus (tropical lineage).
According to previous studies about propagation of tick cell lines infected with the bacteria Anaplasma marginale (Munderloh et al. 1996a, Bastos et al. 2009, Baêta et al. 2015) and Anaplasma centrale (Bell-Sakyi et al. 2015), as well as with Anaplasma phagocytophilum (formerly Ehrlichia equi) (Munderloh et al. 1996b), the L15-B medium was supplemented with low concentration of NaHCO3 as a requirement to guarantee the growth of these pathogens. For successful isolation of those pathogens in tick cell cultures, it was also necessary to reduce the oxygen tension and enhance CO2 tension 100-fold, indicating that some Anaplasmataceae agents are sensitive to oxidation and require elevated CO2 tension for growth (Munderloh et al. 1996b).
In the present study, it was not necessary to supplement the medium with NaHCO3 and the bacteria E. canis (Jaboticabal strain) grew in a regular incubator without CO2 tension as previously described by Singu et al. (2006) in tick cell lines ISE6 from I. scapularis.
The medium used was L-15B supplemented with FBS enriched with iron that showed to be very important to E. canis growth. According to Rikihisa (2010), the proliferation and propagation inclusions of Ehrlichia chaffeensis and Anaplasma phagocytophilum accumulate transferrin receptors in its membrane, favoring the evasion of the immune system and obtaining a continuous supply of nutrients. Iron acquisition by Anaplasmataceae bacteria is dependent on free iron molecules available in the cytoplasm. Although in concentrations (2% and 5%) of FBS enriched with iron we could see infection, the E. canis maintenance in tick cell cultures using L15-B supplemented with 2% of FBS enriched with iron, was satisfactory as well. Levenhagen et al. (2012) analyzed the hole of the cytoskeleton, specifically actin microfilaments and microtubules, and compounds of inositol phospholipid signaling pathway such as phospholipase C, protein kinase, and calcium channels, as well as the hole of iron in the E. canis proliferation in Dh82 cells. These authors observed effective inhibition of E. canis proliferation when infected cells were exposed to the iron chelator, Deferoxamine, showing that this compound affects the soluble iron, not the iron bound to transferrin.
In a previous study, Munderloh et al. (1999) cultivated E. canis in tick cell lines, ISE6, with contaminating erythrocytes and when the erythrocytes become depleted, human fresh and washed erythrocytes were added. The same authors observed that the cells were benefited from the addition of erythrocytes to the culture, because the iron present in these cells plays an important role as a virulence factor in pathogenic bacteria (Munderloh et al. 1999). So our findings reinforce the importance of iron for the growth and maintenance of Anaplasmataceae agents in tick cells and showed that FBS enriched with iron is an alternative to the use of erythrocytes.
The supplementation of the L-15B medium with FBS enriched with iron in infected tick cell cultures under both FBS concentration (2% and 5%) decreased the tick cell growth and improved the E. canis growth, which is fastidious bacteria. Thus, the concentration of 2% FBS enriched with iron showed to be the ideal concentration for growth and maintenance of the bacteria in these cells since the tick cells showed similar conditions under both FBS concentrations. The pathogen grew slowly failing to replicate as quickly as the cells at higher serum concentrations, although the growth was also observed at 5%, as previously observed by Ferrolho et al. (2016).
On the other hand, the uninfected cell cultures of R. sanguineus (tropical lineage), when supplemented with 2–5% of FBS enriched with iron, the cell growth stopped and the cells entered into senescence after 1 month and a half, showing that low FBS concentration causes damage to the cell growth of this cell that requires a high serum concentration (Kurtti et al. 1982).
In our study, we could infect the tick cells and keep the infection in five passages. Although infection of E. canis or other bacteria in primary cultures of tick cells is not commonly used, we have seen that even after thawing, the pathogen remained and was able to infect new cells onto fresh cultures. Probably the primary cultures maintain most of cells resembling the vector. Tick cell lines generally comprise two or more cell types that can be present in varying proportions both at different times within a single culture and at different passage levels (Bell-Sakyi et al. 2007). According to Varma et al. (1975), after subculturing tick cells, some cell types, like large fibroblast-type, eventually disappeared to be replaced by other cell types and they gained or lost chromosomes. These authors observed that cells of R. appendiculatus of 21st subculture showed mixed ploidy. Although most of them were diploid with the male chromosome complement of 21, cells with more and less then this number were also seen.
Regarding the propagation of pathogens in tick cell lines and their continuous cultivation, these attempts have been reported since 70's (Varma et al. 1975). Most of the pathogens, mainly E. canis, have been successfully cultivated in nonvector tick cells, predominantly I. scapularis (Say, 1821) (Bell-Sakyi et al. 2007). The cell lines derived from others tick species, such as Dermacentor variabilis (Say, 1821), Hyalomma anatolicum (Koch, 1844), I. ricinus, I. scapularis, R. appendiculatus, Rhipicephalus evertsi (Neumann, 1897), and R. microplus also showed capacity to support the growth of E. canis (Ferrolho et al. 2016). However, E. canis was successfully propagated in RSE8 cells from R. sanguineus 2 years ago by Ferrolho et al. (2016). Although, RSE8 cells are derived from R. sanguineus ticks, the origin of these ticks is unknown (Ferrolho et al. 2016), so there is no information if the R. sanguineus ticks used to establish the culture belonged to the tropical lineage (able to transmit E. canis) or to the temperate lineage (not able to transmit E. canis). Therefore, the present study reported, for the first time, the propagation of E. canis in primary cell cultures of R. sanguineus tropical lineage (known E. canis vector), from Brazil.
Ganta et al. (2009) observed that in tick cell (ISE6) experimentally infected with E. chaffeensis, the pathogen appeared in phagosomes in both reticulated and dense bodies, whereas in macrophages they appear to grow in synchronized form. The same authors also observed that the reticulated bodies were more pleomorphic and larger in size in tick cells when compared with those observed in macrophages. According to a recent study (Ferrolho et al. 2016), E. canis appeared with double-membrane-bound or loosely packed, disposed, and eventually with electron-dense spots in RSE8 tick cell line, sometimes within the same host cell. Similarly, R. sanguineus (tropical lineage) cells infected by E. canis contained both dense-cored forms and rounded appearance, circled by double membrane, and the reticulated forms.
In conclusion, the present study revealed that primary cell cultures of R. sanguineus (tropical lineage) developed at L-15B medium with 20% of FBS, and in the fifth passage, the cell population doubling time occurred every 57 h. Primary cell cultures of R. sanguineus (tropical lineage) were able to be infected with E. canis. High FBS concentrations enabled the tick cells' survival with the bacteria. The cells of R. sanguineus (tropical lineage) only got infected with E. canis, after they had been kept in L-15B with 2% and 5% FBS fortified with iron. Although in both concentrations (2% and 5%) of FBS enriched with iron we could see infection, the E. canis maintenance in tick cell cultures using L15-B supplemented with 2% of FBS enriched with iron, was satisfactory as well. Primary cell cultures of R. sanguineus (tropical lineage) were also infected with E. canis (strain Jaboticabal) after propagation. Cells derived from R. sanguineus (tropical lineage) were shown to be an alternative for the growth of E. canis in vitro.
Footnotes
Acknowledgments
The authors would like to thank Timothy Kurtti and Urlike G. Munderloh, from the Department of Entomology, University of Minnesota, Minneapolis, for the assistance in preparing the primary tick cultures. Their training in Minneapolis/St Paul in 2007 was fundamental to start the tick cell cultures at the Instituto Butantan. They would like to thank Lesley Bell-Sakyi, from the Pirbright Institute, London, United Kingdom, for providing important information about R. sanguineus s.l. cell lines. The authors are also thankful to Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) (grants no. 2007/57749-2 and no. 2015/26209-9) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq productivity grant, no. 312331/2013-4to DMB-B), for the financial support.
Author Contributions
DMBB performed the study, carried out sampling and laboratory work, and drafted the article; RZM and MRA donated Jaboticabal strain of E. canis and also helped with the improvement of the laboratory work and article; KCMS and JMF helped with the molecular analysis and also drafted the article; LLD, DAF, and ACM helped with all laboratory work and maintenance of the colonies of ticks; PHN performed the TEM pictures and reviewed the article; MMM and MPJS donated R. sanguineus (tropical lineage); MBL reviewed the article critically for important scientific and intellectual content. All authors read, reviewed, and approved the submitted version.
Disclosure Statement
No competing financial interests exist.