
Abstract
Francisella tularensis causes a highly infectious zoonotic disease tularemia. Both Haemaphysalis longicornis and Hyalomma asiaticum are widely distributed in China, but the presence of Francisella and Francisella-like endosymbionts (FLEs) in the two tick species is poorly understood. Therefore, a total of 627 H. longicornis (471 adults and 156 nymphs) and 88 Hy. asiaticum ticks (adults) were collected, of which 88 were from Bole of Xinjiang, 236 from Liaoyang, and 176 from Shenyang of Liaoning, and 215 from Wuhan of Hubei. Notably, five H. longicornis pools from Liaoyang of Liaoning province might have harbored F. tularensis, showing a minimum prevalence of 2.12% (5/236). This study should alert the health department and veterinarians working within the region to prevent and control the emergence of tularemia. After the screening of 16S rRNA and tul4 genes, the results revealed that FLEs were detected in Hy. asiaticum ticks in Bole and in H. longicornis ticks in Liaoyang and Shenyang. Their infection rate was 100% (88/88), 3.39% (8/236 is a minimum), and 8.52% (15/176), respectively. Phylogenetic analyses indicated that the sequence named bole in Hy. Asiaticum from Bole, the sequence named liaoyang1 in H. longicornis from Liaoyang, and the sequence named shanyang1 in H. longicornis from Shenyang shared consistent 16S rRNA sequence, and the difference between Chinese FLEs and the known FLEs was obvious. These findings suggest that this FLE species might be a potentially novel FLE circulating in H. longicornis and Hy. asiaticum from China.
Highlights
Two species of hard ticks were sampled in three different provinces, representing the northwest, northeast, and middle-south regions of China.
Haemaphysalis longicornis ticks from Liaoyang of Liaoning province probably harbored Francisella tularensis.
Phylogenetic analysis revealed the presence of a potentially novel Francisella-like endosymbiont in China.
Introduction
F
Compared with Europe and America, reported tularemia cases are fewer in Asia. In Japan, ∼1400 cases of human tularemia have been reported since 1924 (Ohara et al. 1996). In Korea, only one case of ulceroglandular tularemia (Ahn et al. 1999) and an adolescent patient with atypical pneumonia (Yeom et al. 2015) were reported. Historically, events of tularemia infection in humans have occurred six times in five provinces in China. The earliest reported event of 14 cases was on an isolated island in Heilongjiang province in 1959, and this might have been caused by handling and skinning of dead hares (Kang et al. 1980). The largest outbreak occurred in 1985 in a rabbit-processing plant in Jiaonan County, Shandong Province, where 32 of 36 workers were infected in 12 days (Pang et al. 1987). Since the 21st century, suspicious F. tularensis isolates have been reported in Sichuan, Guizhou, and Anhui provinces one after another. In 2012, a suspected human case was found at Peking University First Hospital in Beijing. The patient's specimens were delivered to the Center for Disease Control and Prevention, China, for the detection of F. tularensis-specific antibodies and analysis of five specific genes. The result indicated that the patient was infected by F. tularensis type B (Wang et al. 2015). In recent years, suspected tularemia cases have occurred frequently. Therefore, it is necessary to find the factors responsible for increasing number of tularemia cases.
The infection can be acquired through wounds, conjunctiva, inhalation of aerosols, or consumption of infected food or water, but the bite of infected arthropods such as ticks should not be ignored, which serve as reservoirs and vectors for F. tularensis (Keim et al. 2007). Ticks are of medical importance owing to their ability to transmit pathogens to humans and animals during feeding. Dermacentor marginatus, D. reticulatus, and Ixodes ricinus ticks of Europe have been reported to harbor F. tularensis (Wicki et al. 2000, Milutinovic et al. 2008, Stanek 2009, Toledo et al. 2009, Franke et al. 2010, Tomanovic et al. 2010). To date, ∼10 genera of ticks and 117 species, and >100 species of Ixodidae have been identified in China (Chen et al. 2010). Among these ticks, Haemaphysalis longicornis and Hyalomma asiaticum are widely distributed in northern and southern China and in other countries of Asia (Teng and Jiang 1991, Chen et al. 2010, Matsumoto et al. 2011, Kang et al. 2013). Understanding the prevalence of F. tularensis in H. longicornis and Hy. asiaticum is significant for risk assessment and monitoring of the spread and emergence of the pathogen in animals and humans.
Furthermore, other members of the Francisellaceae family share high homology with Francisella spp., that is, Francisella-like endosymbionts (FLEs) (Sjöstedt 2005), which are nontransmissible to humans (Niebylski et al. 1997). The implications of this fact in public health are important due to the different pathogenicity of the two organisms. FLEs have a worldwide distribution in both hard and soft ticks (Vun et al. 2000, Machado-Ferreira et al. 2009), including the genera Ixodes, Amblyomma, Dermacentor, and Ornithodoros (Scoles 2004, Machado-Ferreira et al. 2009). The presence of Francisella and FLEs in H. longicornis and Hy. Asiaticum is poorly understood. Hence, this study aimed to investigate the prevalence of F. tularensis and FLEs in the two tick species and the genetic variability of FLEs in Chinese ticks by detecting Francisella-specific nucleic acid.
Materials and Methods
Tick collection
The whole of China can be divided into three parts: the northwest, northeast, and middle-south. A province (i.e., Xinjiang, Liaoning, and Hubei, respectively) was selected from each of the three parts, from where some ticks were collected (Fig. 1). Tick samples were flagged from grass tips or collected from cattle or sheep from four different regions of three provinces. Sampling was carried out in both rural and urban areas of the four cities. Ticks were identified morphologically to the species level using standard taxonomic keys and then verified by analyzing mitochondrial 18S and 12S rDNA sequences (Lu et al. 2013). The ticks were washed in 70% ethanol and distilled water and then processed individually for DNA extraction as described previously (Rijpkemaet al. 1995).
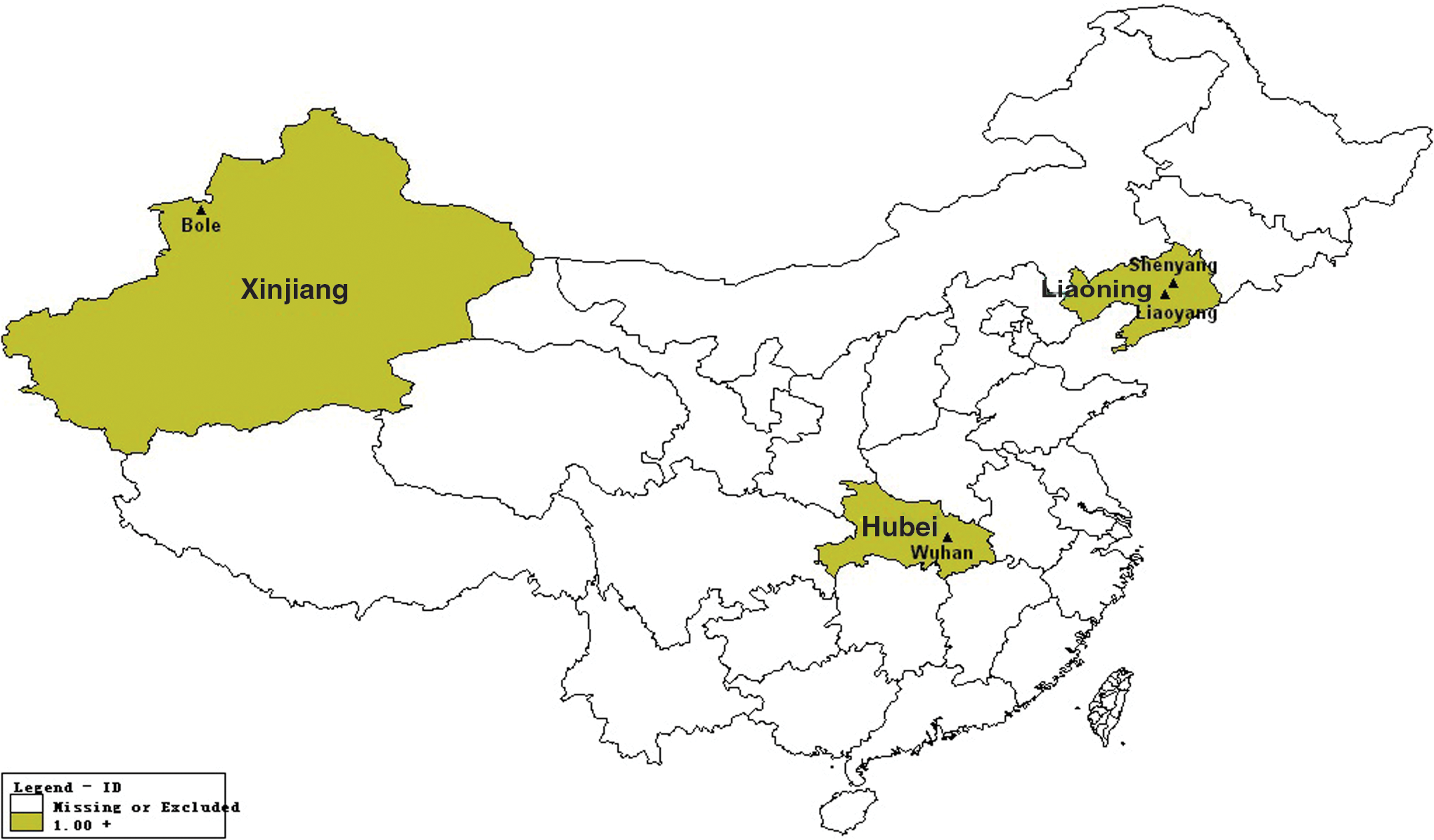
Location of Xinjiang, Liaoning, and Hubei provinces including sampling sites in China. Color images available online at
DNA extraction
A total of 236 H. longicornis ticks from Liaoyang were split into 24 pools, each pool consisting of 9–10 ticks. The DNA of the ticks from Liaoyang was extracted in pools as a unit, and that of the ticks from the remaining three cities in a single tick as a unit. After washing twice with phosphate-buffered saline, a single tick (or ticks from each pool) was homogenized with a mortar and pestle in 0.5 mL (or 1 mL for each pool) of lysis solution with proteinase K. After homogenization, the suspension was incubated at 4°C for 1 h and centrifuged at 2500 g for 5 min. The upper fraction was collected and the DNA was extracted from an individual tick or each pool using the DNeasy Tissue Kit according to the manufacturer's instructions.
PCR and sequencing
All extracted DNAs were tested with the 16S rRNA gene-based Francisella-specific conventional PCR, using the modified primer pairs based on previous reports (Forsman et al. 2000, Dergousoff and Chilton 2012). Forward primer Ft16S-F: 5′-TACCAGTTGGAAACGRCTGT-3′ and reverse primer Ft16S-R: 5′-TGCGGGACTTAACCCAACAT-3′ were used, amplifying a 949-bp fragment. A final reaction volume of 20 μL comprised 2 × mix buffer of 10 μL, forward and reverse primers (each 0.6 μL), and template of 8.8 μL. The amplification program consisted of 95°C for 5 min, followed by 30 cycles of 95°C for 1 min, 59°C for 1 min, and 72°C for 1 min, and a final extension at 72°C for 10 min. F. tularensis ssp. holarctica (live vaccine strain [LVS], NCTC 10857) served as a positive control, and distilled water was used as a negative control during nested PCR of 16S rRNA. The resulting amplicons were analyzed by agarose gel electrophoresis. Each reaction setup was performed in separate rooms using exclusive pipettes and tips to avoid contamination.
All samples were further subjected to conventional PCR based on the tul4 gene specific for the genus Francisella, using the primer pair FT393/FT642 (Long et al. 1993), amplifying a 250-bp fragment of the gene coding the 17-kDa lipoprotein (tul4) of Francisella spp. as described in a previous study (Karhukorpi and Karhukorpi 2001). The resulting amplicons were also analyzed by agarose gel electrophoresis. A negative control (distilled water) instead of tick DNA template in the PCR master mix and a positive control (DNA from LVS) were included during each reaction.
DNA sequencing of the PCR products was performed using 377 gene sequencers (Applied Biosystems, USA) at Tianyi Huiyuan Biological Engineering Technology and Services Co., Ltd. (Beijing, China) using the Sanger sequencing method. Nucleotide sequences generated in this study have been deposited in the GenBank database under accession numbers KX808676–KX808679, KX811540–KX811542, and KX811545.
Phylogenetic tree analyses
Nucleic acid databases of Francisella were searched using the BLASTN program in GenBank. The reading errors of the chromatograms were corrected, and alignments (16S rRNA, 949-bp; tul4, 250-bp) of the PCR amplicons of the two genes recovered through this study were performed with existing reference sequences in GenBank using ClustalW (default parameters) as implemented in the MEGA program (version 6.06) (Tamura et al. 2013). The primer sequences were removed from the alignment before phylogenetic analyses.
The best-fit evolutionary model for all sequence alignments was determined using the jModel Test version (Posada 2008). The Hasegawa–Kishino–Yano nucleotide-based model was found to be the best-fit model for 16S rRNA and tul4 genes. Phylogenetic trees were then estimated using the maximum likelihood method implemented in PhyML (version 3) (Guindon et al. 2010). Moreover, 100 bootstrap replicates were obtained under the same procedure, and bootstrap values >50% support were indicated at the node to estimate support for individual nodes. All trees were mid-point rooted for clarity.
Results
Collection of ticks
Figure 1 shows a map of China and three provinces comprising four collection sites. The regions of the three provinces are shown in light brown and names of provinces are highlighted in light blue. The small black triangles represent locations of collection sites of the three provinces, and the names of the collection sites are marked in black font. The locations of the four sampling sites represent the northwestern, northeastern, and mid-southern regions of China, respectively, wherein the two sampling sites are from Liaoning province.
A total of 627 H. longicornis and 88 Hy. asiaticum ticks were collected after morphological examination and sequence analysis of mitochondrial 18S and 12S rDNA sequences as described in a previous study (Lu et al. 2013). The source of all ticks was as follows: 88 from Bole of Xinjiang, 236 from Liaoyang, 176 from Shenyang of Liaoning, and 215 from Wuhan of Hubei (Table 1). The range of collection time was from May 2011 to May 2013. Moreover, 425 ticks were sampled from domestic animals (sheep or cattle) and 290 from grassland. Of the 715 ticks collected, 78% were adults (559/715) and 22% were nymphs (156/715).
Composition of the Two Tick Samples and Detection of Francisella tularensis and Francisella-Like Endosymbionts
Detection of Francisella 16S rRNA gene
The total tested number of two tick species was 503 for 16S rRNA gene, including 479 single ticks and 24 pools. All 88 Hy. asiaticum ticks from Bole gave positive results in this PCR and shared identical 16S rRNA sequences named bole (GenBank acc. no. KX808676). The infection rate of the Hy. asiaticum ticks was 100% (88/88). For H. longicornis from Liaoyang, 13 out of 24 pools gave positive results and had 2 variants named liaoyang1 (8/13, acc. no. KX808677) and liaoyang2 (5/13, acc. no. KX808678). The pooled infection rate of the H. longicornis ticks was 54% (13/24). For H. longicornis from Shenyang, 15 out of 176 ticks gave positive results and 1 had variants named shenyang1 (acc. no. KX808679). The infection rate of H. longicornis ticks from Shenyang was 8.5% (15/176). For H. longicornis ticks from Wuhan, none were positive for the 16S rRNA gene (Table 1).
Detection of Francisella tul4 gene
The total tested number of the two tick species was also 503 for tul4 gene and comprised 479 single ticks and 24 pools. For Hy. asiaticum from Bole, 59 out of 88 ticks gave positive results and also shared identical tul4 sequences named bole 59 (KX811540, Table 1). Notably, all 24 pools of H. longicornis ticks from Liaoyang gave positive results and also shared identical tul4 sequences named liaoyang34 (KX811541). For H. longicornis from Shenyang, only 4 out of 176 ticks gave positive results for tul4 gene, and simultaneously these ticks gave positive results for 16S rRNA gene. The tul4 sequences of four ticks had one variant named SY54 (KX811542). For H. longicornis from Wuhan, only 1 out of 215 gave positive result. The tul4 sequence of this tick had one variant named CJM18 (KX811545).
Phylogenetic relationship based on 16S rRNA gene
This study aimed to evaluate the presence of F. tularensis and FLEs in the ticks collected from three different geographical areas in China. A phylogenetic tree based on 16S rRNA gene sequences of the representative tick specimens, F. tularensis and FLEs, in and outside China is displayed in Figure 2. It was indicated that 5 (named liaoyang2, KX808678) of 24 H. longicornis pools from Liaoyang cluster were within the sequences of F. tularensis. In the case of Francisella-positive H. longicornis tick samples, each represented a minimum prevalence (with only one infected tick per pool) of 2.12% (5/236).
Phylogenic tree inferred from the 16S rRNA sequences of Francisella strains, FLEs, and tick samples. The scale bar represents the number of nucleotide substitutions per site. The new sequences provided by this study are marked in red. N means the number of same sequences. FLEs, Francisella-like endosymbionts; LVS, live vaccine strain. Color images available online at
In contrast, the remaining three sequences (bole, liaoyang1, and shenyang1) clustered into another group with sequences of FLEs. In the 16S rRNA tree, bole in Hy. asiaticum from Bole, liaoyang1 in H. longicornis from Liaoyang, and shanyang1 in H. longicornis from Shenyang shared consistent 16S rRNA sequence. At node value 79, these three sequences clustered on a single clade and formed a lineage with 16 FLEs from the rest of the world.
Phylogenetic relationship based on tul4 gene
A phylogenetic tree based on tul4 gene sequences of the representative tick specimens, F. tularensis and FLEs, in and outside China is displayed in Figure 3. In the tul4 phylogenetic tree, all 24 H. longicornis pools from Liaoyang shared an identical sequence (acc. no. KX811541) and clustered together with the sequences of F. tularensis. The 24 tul4-positive pools comprised the aforementioned five 16S rRNA-positive pools. It was noted that, except ticks from Liaoyang, bole59, SY54, and CJM18 also clustered within the sequences of F. tularensis. However, in the 16S rRNA phylogenetic tree, 16S rRNA sequences of these ticks were not clustered together with F. tularensis.

Phylogenic tree inferred from the tul4 sequences of Francisella strains, FLEs, and tick samples. The scale bar represents the number of nucleotide substitutions per site. The new sequences provided by this study are marked in red. N means the number of same sequences. Color images available online at
Discussion
In this pilot study, a total of 391 ticks and 24 pools of H. longicornis and 88 Hy. asiaticum ticks from three distinct provinces in China were screened for the presence of F. tularensis or FLEs. This screening revealed that amplicon sequences of five H. longicornis pools from Liaoyang of Liaoning province were consistent with the presence of F. tularensis, showing a minimum prevalence of 2.12% (5/236). In addition, FLEs were detected in Hy. asiaticum of Bole and in H. longicornis of Liaoyang and Shenyang from two distinct provinces. Their infection rate was 100% (88/88), 3.39% (8/236 is a minimum), and 8.5% (15/176), respectively.
The 16S rRNA tree is quite similar to the phylogenetic framework of a work based on whole genome (Ahlinder et al. 2012). In this study, five 16S rRNA gene sequences of the Francisella-positive H. longicornis tick samples from Liaoyang clustered with F. tularensis. Also, their tul4 gene sequences were positive for Francisella, implying that they belonged to F. tularensis. Two publications reported the presence of F. tularensis in H. longicornis and other tick species in China. An investigation was carried out to test eight pathogens, including F. tularensis in tick species collected from the southern, central, and northeast regions of China. The results indicated that Borrelia carolinensis and B. bissettii were detected in H. longicornis, whereas no F. tularensis was found (Yu et al. 2016). Zhang et al. (2008) reported D. silvarum and I. persulcatus from China harboring F. tularensis in the natural environment, and the prevalence averaged 1.98%. A Japanese study detected the presence of Anaplasma, Bartonella, Borrelia, Ehrlichia, and Rickettsia in seven tick species by Batch Learning Self-Organizing Map analysis. The results showed that Francisella was detected in I. ovatus, I. persulcatus, I. ricinus, and Amblyomma testudinarium, but not in H. longicornis, A. variegatum, and H. formosensis (Nakao et al. 2013). This novel study reported the presence of F. tularensis in H. longicornis ticks from Liaoyang of Liaoning province, China. This should alert the health department and veterinarians working within the region on the need to prevent and control the emergence of tularemia.
The distribution and prevalence of FLEs in tick species that transmit tularemia are largely unknown, as only a few studies have been performed (Sun et al. 2000, Scoles 2004, Kugeler et al. 2005). In this study, FLEs were detected in both H. longicornis and Hy. asiaticum ticks with a higher infection rate, and their distribution was implicated in two targeting provinces located in the northwest and northeast of China, but not in Hubei province. In the 16S rRNA tree, the sequence of FLEs was identical in Hy. asiaticum from Bole and H. longicornis from Liaoyang and Shenyang. The same FLE species was shown to be circulating in the two tick species. Regarding a host species-linked evolution of certain FLE species, this point of view was challenged by the present finding, but was the same as shared by another study (Scoles 2004). Moreover, these FLE species come from two distinct geographical areas (Xinjiang and Liaoning provinces) located in the northwestern and northeastern China, respectively. The result revealed some biological characteristics of these FLE species that they might prefer to live in a colder area. The FLE species from China formed a lineage with 16 FLEs and shared an identical sequence that has not been found in any other country. Chinese FLEs alone clustered together into a group, in which obvious differences appeared between them and the known FLEs. These findings suggested that this FLE species might be new FLEs circulating in H. longicornis and Hy. asiaticum from China. Several lines of research were performed on screening the presence of tick-borne pathogens in Hy. asiaticum ticks from China (Xia et al. 2011, Zhang et al. 2013, Li et al. 2015), but F. tularensis was not included. This novel study detected the presence of FLEs in Chinese H. longicornis and Hy. asiaticum ticks, confirming the presence of these bacteria and the need to take this into account in ticks and environmental specimens.
In the tul4 phylogenetic tree, bole59, liaoyang34, SY54, and CJM18 clustered within the sequences of F. tularensis. Liaoyang34 shared an identical sequence in 24 pools in H. longicornis ticks from Liaoyang, and 16S rRNA sequence of only 5 out of 24 pools was positive for F. tularensis. In the obtained ticks, for tul4 gene positive bole59 and SY54, except CJM18, 16S rRNA genes were positive for FLEs. It follows that bole59 and SY54 should belong to FLEs instead of F. tularensis. This might have led to misidentification in distinguishing between F. tularensis and FLEs by tul4 gene sequence. Similar results were obtained during the examination of ticks from Bulgaria, where only 6 out of 12 16S rRNA gene-based PCR-positive FLEs were positive using PCR assay of the tul4 genes (Sjöstedt et al. 1990, Ivanov et al. 2011). Moreover, two different standard tul4 PCR assays have also been shown to cross-react with the FLEs in Dermacentor species (Niebylski et al. 1997, Scoles 2004). This might necessitate progressive adaptation of molecular tools, allowing accurate identification of FLE species in the near future.
Conclusion
The results obtained in this study indicate that H. longicornis from Liaoyang of Liaoning province probably harbored F. tularensis, showing a minimum prevalence of 2.12%. This novel study reported the presence of F. tularensis in H. longicornis ticks from China, which should alert the health department and veterinarians working within the region to prevent and control the emergence of tularemia. Also, it should be appreciated that FLEs were detected in Hy. asiaticum of Bole and in H. longicornis of Liaoyang and Shenyang from two distinct provinces. Phylogenetic analyses revealed that Chinese FLEs alone clustered together into a group, and obvious differences appeared between them and the known FLEs. These findings suggest that this FLE species might be a new FLE circulating in H. longicornis and Hy. asiaticum from China.
Footnotes
Acknowledgments
The authors thank Prof. Jiang Tian and Dr. Wen Wang for their assistance in collecting ticks. Financial support for this study were provided by National Natural Science Foundation of China (grant no. 81473032) and China Mega-Project for Infectious Disease (no. 2012ZX10004219).
Authors' Contributions
W.Y.H. wrote the article and performed the experiments. M.L.L., Z.J.B., and X.L.X. designed the experiments. S.Y.W., W.Z.J., Z.J.Y., and P.Y. collected the samples. All authors read and approved the final article.
Author Disclosure Statement
No competing financial interests exist.