
Abstract
Bartonella is a species-rich bacterial genus that infects a wide variety of wild and domestic animals, including rodents. Despite high levels of murid rodent diversity in Africa, associated Bartonella prevalence and diversity remains understudied, particularly within the southern African subregion. To address this, we sampled endemic four-striped mice, Rhabdomys pumilio, from three rural and two urban localities in the Western Cape Province, South Africa. PCR screening and multilocus sequence analysis inclusive of five genome regions (gltA, nuoG, ribC, rpoB, and ITS), were respectively used to evaluate Bartonella status and diversity in these synanthropic rodent populations. An overall infection rate of 15% was recovered, ranging from 0% for an urban locality to 36.4% for a rural locality, consistent with the higher flea abundance recorded at the latter sites. Nucleotide sequencing and phylogenetic analyses confirmed the presence of three distinct Bartonella lineages (I–III), with lineages II and III grouping with bartonellae previously detected in R. pumilio from nature reserves in the Free State Province of South Africa, and lineage I being novel and sister to Bartonella strains identified previously in Micaelamys namaquensis. Our results indicate significant landscape effects on infection rates, highlight differential PCR assay performance, and identify three host-associated Bartonella lineages in Rhabdomys from South Africa.
Introduction
Bartonellae are fastidious, Gram-negative α2-proteobacteria (Houpikian and Raoult 2001) that infect a wide range of mammalian hosts, including rodents, insectivores, carnivores, lagomorphs, primates, ungulates, and humans (Chomel and Kasten 2010). These bacteria typically infect erythrocytes of vertebrate reservoir hosts and can induce pathology in multiple organs (Young et al. 2014). Transmission among vertebrate hosts is facilitated by blood-sucking arthropod vectors, such as fleas, ticks, mites, and sand flies, but fleas, in particular, are considered major vectors among rodent hosts (Billeter et al. 2008, Tsai et al. 2011).
At least 34 species and subspecies of Bartonella are currently recognized with the majority of these (>80%) recorded in rodents (Jiyipong et al. 2014). The ability of rodents to invade anthropogenically transformed areas where they can attain pest status, and their role as reservoir hosts for a wide range of infectious disease agents, including Bartonella, underscore the importance of assessing the bacterial infection status of rodents across different habitat types, and particularly within commensal settings where contact opportunities are high (Bastos et al. 2005, Berglund et al. 2010).
Members of the genus Rhabdomys are relatively small (40–60 g), widely distributed rodents, occurring throughout southern and East Africa (Skinner and Chimimba 2005). Initially considered a monotypic genus comprising between 14 and 20 Rhabdomys pumilio subspecies, the genus has undergone a number of revisions, with molecular data now suggesting the presence of at least two southern African species, R. pumilio and Rhabdomys dilectus, and two R. dilectus subspecies (Rambau et al. 2003, Castiglia et al. 2012, Du Toit et al. 2012, Ganem et al. 2012). Areas of sympatry in South Africa have been inferred from environmental niche modeling (Meynard et al. 2012) and confirmed through molecular characterization (Le Grange et al. 2015).
R. pumilio is a broad-niche, peridomestic rodent that can adapt to and benefit from anthropogenic habitat change (Froeschke et al. 2013, Van der Mescht et al. 2013). In line with the generalist nature of this rodent, it is host to a diverse array of parasitic species. In the Western and Northern Cape Provinces of South Africa more than 30 ectoparasite species, including ixodid ticks, mesostigmatid and trombiculid mites, fleas and anoplurid lice, have been recorded from R. pumilio (Matthee et al. 2007, 2010, Matthee and Ueckermann 2008, Matthee and Krasnov 2009, Froeschke et al. 2013), with some of the recorded fleas (e.g., Chiastopsylla rossi and Dinopsyllus ellobius) being known vectors of zoonotic disease agents, such as Yersinia pestis (Ingram 1927). More recently, Van Der Mescht and Matthee (2017) reported the presence of at least 14 flea species from R. pumilio, viz. Chiastopsylla capensis, Chiastopsylla carus, Chiastopsylla mulleri simplex, Chiastopsylla pitchfordi, Chiastopsylla quadrisetis, C. rossi, Ctenophthalmus calceatus, D. ellobius, Dinopsyllus tenax, Hysophthalmus temporis, Listropsylla agrippinae, Listropsylla fouriei, Xenopsylla brasiliensis, and Xenopsylla eridos.
A prior study of bartonellae in terrestrial small mammals from nature reserves in the Free State Province of South Africa revealed high levels of infection and diversity in the 10 mammalian host species evaluated (Pretorius et al. 2004). Although just 9 of the 86 animals were R. pumilio specimens, characterization of the citrate synthase (gltA) gene revealed that the three Bartonella-positive Rhabdomys specimens harbored two novel lineages. The uniqueness of these lineages and relatively high Rhabdomys infection rate (44%) prompted the current expanded assessment of Bartonella prevalence and diversity in R. pumilio sampled from rural and urban settlements in the Western Cape Province of South Africa. Using multilocus screening in combination with phylogenetic analyses we were able to obtain refined estimates of infection, across these modified habitat types and enhanced taxonomic placement of the Bartonella lineages present in Rhabdomys.
Materials and Methods
Rodent sampling
A total of 80 R. pumilio individuals were sampled from five localities in the Western Cape Province as part of a previous study (Froeschke and Matthee 2014). Two of the five localities were urban settings (Franschhoek and Somerset West) and three occurred within rural landscapes (Franschhoek, Wellington and Gordon's Bay). Line transects, consisting of Sherman-like live traps were placed ∼10 meters apart and baited with a mixture of peanut butter and oats. R. pumilio individuals were euthanized with 2–4 mL sodium pentobarbitone (200 mg/kg), depending on individual weight, as approved by the Ethics Committee of Stellenbosch University [ref no. 2006B01007 and SUACUM11-00004(p)] and under collection permits issued by Cape Nature (ref no. 317/2003, 360/2003, AAA004-00221-0035). Body measurements and weight were recorded for each animal, and the ectoparasites were removed by pelage brushing and stored in 70% ethanol. Fleas were mounted using standard methods and identified to species level (Segerman 1995) by a single investigator (L.v.d.M.).
Genetic characterization
DNA extracted from frozen Rhabdomys heart tissue with the High Pure PCR Template Preparation Kit (Roche) was screened for Bartonella genome presence as previously described using primer sets targeting the citrate synthase (gltA), NADH dehydrogenase gamma subunit (nuoG), riboflavin synthase (RibC), RNA polymerase subunit B (rpoB), and the 16S–23S ribosomal RNA intergenic spacer (ITS) gene regions (Supplementary Table S1. As fleas were mounted for identification purposes, they were not evaluated for Bartonella genome presence.
Samples were considered positive based on at least two independent amplifications of one or more target gene regions and on nucleotide sequence verification of Bartonella presence. The latter was achieved by purifying amplicons of the correct size with the High Pure PCR Product Purification Kit (Roche), and performing Sanger cycle sequencing with Big Dye Terminator Cycle Sequencing Ready Reaction Kit (PerkinElmer), with each of the external PCR primers, in separate reactions. Reactions were purified, denatured, and submitted to the core Sanger sequencing facility at the University of Pretoria.
Phylogenetic analyses
Individual Bartonella gene datasets complemented with homologous, reference sequence data from the GenBank database (
Statistical analyses
A stepwise Akaike Information Criterion (AIC) analysis was conducted to determine whether host sex, landscape type (urban vs. rural), and load of each flea species or total host flea load significantly explained the variation in Bartonella status providing the best-fit predictive model. A binomial logistic regression was conducted on the best-fit model to determine whether any of the predictors had a significant effect on the log probability of Bartonella status. A Fisher's exact test, to account for limited flea species data, and a Pearson's chi-squared test (with Yates' continuity correction) were conducted to test whether Bartonella status is dependent on the species or sex of fleas, respectively. All analyses and assumption testing were conducted in the Rstudio interface of R v3.4.3 (R Core Team 2017).
Results
Bartonella amplification ranged from 6.5% to 13.8% (Table 1), across the different gene regions. Based on two or more independent, sequence-confirmed Bartonella genome amplifications, an overall Bartonella infection rate of 15% was recovered (Table 1). Whereas the gltA and ribC primer sets each detected 10 positive individuals, the nuoG, rpoB, and ITS assays identified 11, 8, and 5 Bartonella-positive animals, respectively.
Locality, Geographical Coordinates, Sample Size, and Bartonella PCR Assay Detection Rates in Rhabdomys pumilio Sampled from Rural and Urban Sites in the Western Cape Province, South Africa
ITS, intergenic spacer.
Sequence data generated in this study were submitted to GenBank under accession numbers indicated in Supplementary Table S2. These data, when complemented with reference strain sequences in the GenBank database, were used to infer individual gene phylogenies (Supplementary Figs. S1 and S2) to confirm the consistency of phylogenetic placement across the different gene regions. In addition, samples containing discernible levels of coinfections across the different gene regions characterized (Supplementary Table S3) were excluded before concatenation. As most samples were coinfected, this ultimately restricted enhanced phylogenetic placement of lineages within the broader genus phylogeny to just two samples, FHC6 and GB17, respresentative of lineages I and III, respectively. GltA and ribC gene phylogenies each recovered three discrete lineages (denoted I–III in the gltA gene tree; Fig. 1A), of which two (II and III) grouped with bartonellae previously identified in R. pumilio from the Free State Province of South Africa (Pretorius et al. 2004). In contrast, lineage I was novel and sister to bartonellae strains previously identified in Micaelamys namaquensis from Gauteng Province, South Africa (Brettschneider et al. 2012). A number of close matches (≥97% identity) were identified for the gltA gene through nucleotide Blast searches against the GenBank database (
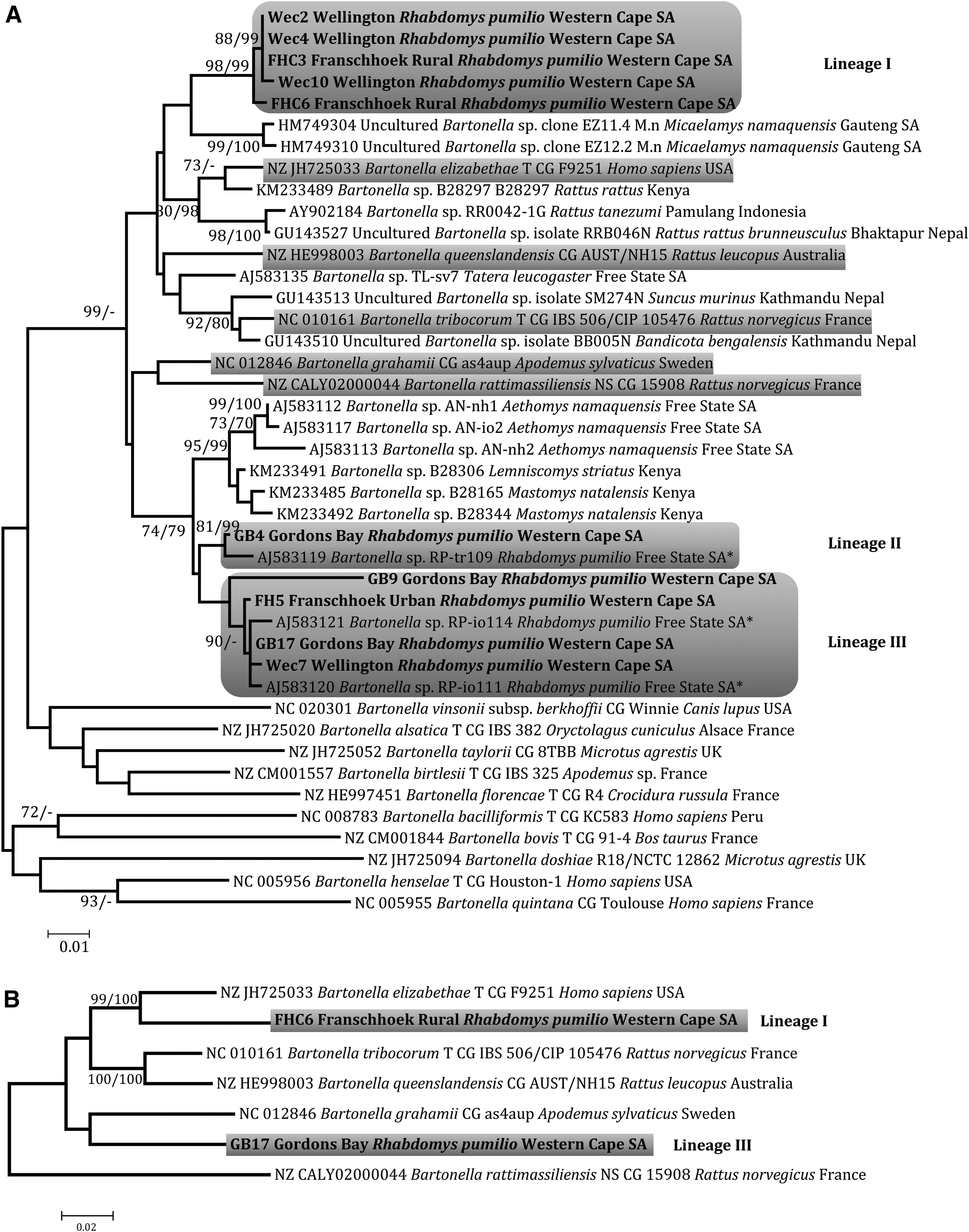
ML trees inferred using:
Three flea species, C. rossi, D. tenax, and L. agrippinae, were recorded from R. pumilio at the five localities (Table 2). Although abundance varied between localities, two species (C. rossi and L. agrippinae) predominated at all sites. Tick larvae and nymphs (Rhipicephalus, Haemaphysalis, and Ixodes) as well as mesostigmatid mites (Laelaps giganteus) were also present.
Abundance and Prevalence of Flea Species Recorded from Rhabdomys pumilio at Five Localities in Rural and Urban Landscapes in the Western Cape Province, South Africa
Under the binomial logistic regression; landscape type (urban vs. rural) had a significant effect on the log probability of Bartonella occurrence (χ 2 = 8.66, df = 1, p = 0.003245) with rodents from the rural landscape being significantly more likely to have a higher infection rate compared with those sampled from the urban landscape (p = 0.02552). However, the best-fit predictive model did not contain load of each flea species or total host flea load, thus our flea data had no significant role in explaining the variation in Bartonella prevalence. Additionally, neither the Fisher's exact test, nor the Pearson's chi-squared test (with Yates continuity) were significant, indicating that Bartonella status was independent of flea species composition and sex, respectively.
Discussion
The overall Bartonella infection rate in R. pumilio samples collected in the present study was 15%, ranging from 0% (for Somerset West) to 36.4% (for Wellington) by locality, and from 6.5% (for ITS) to 13.8% (for nuoG) in the individual gene assessments (Table 1). As frozen heart samples, rather than spleen, were used in this study, it should be noted that this likely represents an underestimation of the true infection rate. The marked variation in Bartonella genome detection capabilities of the assays precluded characterization of all gene regions for the three lineages detected in Rhabdomys from the western Cape and highlights the importance of using multiple assays in parallel to ensure accurate estimates of Bartonella prevalence. Despite variable assay performance, the individual gene phylogenies gltA (Fig. 1A) as well as the ribC, rpoB, and ITS (Supplementary Fig. S1) consistently recovered two of the three Bartonella lineages (I–III) in R. pumilio. However, the nuoG marker (Colborn et al. 2010) identified two additional lineages (Supplementary Fig. S2). These results are at odds with the more widely used gene markers that have proven valuable for delineating rodent-associated Bartonella species (La Scola et al. 2003, Pretorius et al. 2004), suggesting that while the nuoG marker is valuable for detecting Bartonella genome presence in clinical specimens, its phylogenetic utility is less clear.
Using the prescribed p-distance cutoffs for the gltA and rpoB gene regions (<96% and <95%, respectively; La Scola et al. 2003), it was confirmed that six positive specimens harbored a strain that is sister to the zoonotic B. elizabethae species complex, and which is denoted lineage I in this study. The remaining R. pumilio individuals harbored one of two lineages (denoted II and III in this study) previously identified in R. pumilio individuals (Pretorius et al. 2004) sampled from a natural setting more than 800 km north of our study site. Of interest is that the gltA p-distance phylogeny placed one sample from lineage III (GB9) outside the La Scola et al. (2003) species delineation criteria, whereas p-distances for other gene regions placed the same sample well within the proposed cutoff values (La Scola et al. 2003). Hence, in accordance with Pretorius et al. (2004), we suggest that the cutoff values for individual gene regions should not be applied too strictly and should also be guided by nodal support values confirming monophyly of the novel lineage.
On the basis of the five-gene multilocus sequence analysis (Fig. 1B), two of the three lineages (I and III) present in Rhabdomys appear to be novel with lineage III corresponding to a strain previously detected in Rhabdomys (Fig. 1A; Pretorius et al. 2004) and lineage I being closely related to B. elizabethae. This warrants further investigation as strains of the latter Bartonella species complex are zoonotic and ubiquitous. Furthermore, whereas lineage I predominated at the three rural sampling sites evaluated in this study (Table 1), it was not recorded in Rhabdomys previously sampled from natural landscapes (Pretorius et al. 2004).
In the present study, the Bartonella infection rates in R. pumilio varied between habitat types with rural landscapes generally having higher levels of infection than urban landscapes. Despite the relatively small sample size and similar densities of R. pumilio hosts in both landscape types (Froeschke and Matthee 2014), statistically significant differences were observed between urban and rural localities. The highest infection rate was recorded at the rural locality of Wellington (36.4%), whereas at the urban locality of Somerset West there was an absence of Bartonella. The former infection rate is comparable to the 44% infection rate in R. pumilio sampled from natural landscapes in the Free State Province and reported by Pretorius et al. (2004). This, together with the observation that Bartonella infection rates were higher at the Franschhoek rural locality (25%) compared with the Franschhoek urban locality (7.7%) suggests that land usage type and the associated degree of anthropogenic modification appears to be a strong driver of Bartonella prevalence.
Ectoparasites, especially fleas, are known vectors of Bartonella species among rodents (Billeter et al. 2008, Tsai et al. 2011) and it is therefore anticipated that higher infestation rates would be associated with higher Bartonella infection rates. However, our statistical analyses suggest that this is not the case for the Western Cape population of R. pumilio evaluated in this study.
Conclusions
From the limited number of studies of Bartonella in murid rodents sampled from natural settings in South Africa (Pretorius et al. 2004, Brettschneider et al. 2012), it is clear that the high levels of regional murid rodent biodiversity (Skinner and Chimimba 2005) reflect similarly high levels of Bartonella species diversity and infection rates. While studies that determine overall infection and diversity for multiple rodent host species at a single sampling site are valuable, focused single-host studies provide a means for unraveling Bartonella transmission dynamics. In an assessment of M. namaquensis, an indigenous murid rodent that can attain pest status, seasonal variation in Bartonella infection was detected by Brettschneider et al. (2012), whereas in the current study a landscape effect was discernible, despite the small sample size. Both studies underscore the value of expanded, single-host species investigations in reaching a better understanding of factors influencing rodent Bartonella ecology in southern Africa.
Footnotes
Acknowledgments
L.H. and D.K. were supported by Centers for Disease Control and Prevention (CDC) Cooperative Agreement (5 NU2GGH001874-02-00) postgraduate bursaries and G.F. by a free-standing postdoctoral bursary from the National Research Foundation (NRF) of South Africa. The research was conducted with financial support from the University of Pretoria's Animal and Zoonotic Diseases Institutional Research Theme (AZD-IRT), CDC Co-Ag 5 NU2GGH001874-02-00, the M3B2 DST/NRF SARChI Chair and through individual (ADSB, SM) and facility (no: UID78566) NRF grants. The contents are solely the responsibility of the authors and do not necessarily represent the official views of the CDC, nor the NRF.
Author Disclosure Statement
No conflicting financial interests exist.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.