
Abstract
Tick-borne encephalitis virus (TBEV) can cause fever, headache, neurological disorders, and/or peripheral flaccid paralysis; therefore, it is a major threat to public health. A rapid, sensitive, and simple method for detecting anti-TBEV antibodies is needed urgently to determine infection and for vaccine evaluation. Here, a luciferase-based immunocomplex assay system (Luc-IC) was developed to detect TBEV antibodies. The system is based on a reporter Nano luciferase (NLuc) that is co-expressed as a fusion protein with viral envelope domain III (ED3) in COS7 cells. The cell supernatant was used directly to detect antigen without the need for a purification step. This simple procedure effectively improved the sensitivity of the assay. Sera from 50 patients with an acute tick-borne encephalitis infection were tested to determine the sensitivity of the NLuc-IC assay. Furthermore, 62 sera from individuals infected with Japanese encephalitis virus, West Nile virus, yellow fever virus, dengue virus, or Zika virus were also tested to determine specificity. The results demonstrated that the assay was 100% sensitive and 100% specific for TBEV antibodies. Thus, this very simple NLuc-IC assay is potentially useful for rapid and accurate diagnosis of TBEV infection in both humans and animals.
Introduction
Tick-borne encephalitis virus (TBEV), which belongs to the genus Flavivirus within the family Flaviviridae, can cause severe meningitis and encephalitis. Symptoms include fever, headache, neurological disorders, and/or even peripheral flaccid paralysis (Mansfield et al. 2009). TBEV infects humans through tick bites; indeed, there are at least 14,000 human cases of encephalitis in Russia, China, and European countries annually. Thus, the virus poses a serious threat to public health.
Rapid testing and diagnosis of possible human TBEV infection is an important public health task. Presently, laboratory diagnosis of TBEV includes virus isolation, detection of virus-specific antibodies, and detection of viral genomic sequences using nucleic acid amplification techniques. Virus isolation is the gold standard test for TBEV infection; however, it requires biosafety level 3 containment facilities and places technicians at a high risk of infection.
Presently, for serological TBEV detection, the neutralization test (NT), immunoglobulin G (IgG) enzyme-linked immunosorbent assay (ELISAs), and immunofluorescence assay (IFA) have been well developed. NT is the most specific assay available; however, it needs biosafety level 3 containment facility and puts the technicians at a high risk of becoming infected with TBEV. IFA assays use the TBEV-infected cell culture as the detection antigen and can reveal high sensitivities, but showed a high degree of cross-reactivity with other flaviviruses, such as Japanese encephalitis virus (JEV), dengue virus (DENV), yellow fever virus (YFV), West Nile virus (WNV), or Zika virus (ZIKV). ELISA is also widely used for serological detection. The mainly used diagnostic antigen for TBEV infection is the inactivated viral cultures. Cross-reactions and risk of being infected also exist. As for recombinant proteins, the process of recombinant protein preparation could eliminate the biosafety requirement and improve the specificity of diagnostic tests (Morozova et al. 2014). The envelope glycoprotein (E protein) of TBEV comprises 496 amino acids; this is the antigen commonly used in ELISA. Assays based on E protein showed sensitivity for TBEV comparable with that of native viral cultures; however, cross-reactions with other flaviviruses are possible (Obara et al. 2006, Klaus et al. 2011, Larsen et al. 2014, Morozova et al. 2014). Domain III of the envelope protein (ED3) is implicated in receptor binding and is a target for TBEV-specific neutralizing antibodies. Previous studies indicated that a sero-complex assay based on ED3 is specific for tick-borne encephalitis (TBE) infection, with no cross-reactivity with other flavivirus-positive sera (Allison et al. 1999, Peter et al. 2008, Ludolfs et al. 2009, Chavez et al. 2010, Rockstroh et al. 2011, Schmitz et al. 2011); however, the assay lacks sensitivity.
Early and specific diagnosis of TBE is highly desirable for both detection and surveillance of TBEV. At present, no commercial assay kits for TBEV infection are available in China. ELISAs and IFAs are the tests most commonly used by hospitals to diagnose and monitor immune responses to TBEV infection.
Here, we aimed to develop ED3 as a potential diagnostic antigen and establish a rapid, sensitive, and specific luciferase immune complex assay system for detecting anti-TBEV antibodies. The assay uses the ED3 region of the TBEV envelope protein as the antigen. The assay detects immune complexes formed by human anti-TBEV antibodies and a luciferase-labeled ED3 antigen. The accuracy (specificity and sensitivity) of the luciferase-based immunocomplex assay system (Luc-IC) assay was evaluated and compared with that of an indirect IFA and an indirect ELISA (ED3-ELISA) based on the ED3 antigen.
Materials and Methods
Clinical serum samples
Samples of serum taken from 50 clinical TBE cases were collected. All cases were determined according to symptoms of fever, headache, disturbance of consciousness or hypnosis, epidemiological inquiry of tick biting, and detection of TBEV-specific IgM and IgG antibodies by IFA. In addition, 62 serum samples from subjects not exposed to TBEV or to a TBE vaccine were used as negative controls to determine the specificity of the new assay; these included 10 healthy samples, 15 clinical dengue fever cases, 28 cases of JEV infection, three cases of ZIKV infection, three sera from WNV-infected rabbits, and three sera from YFV-infected mice.
Production of ED3 antigen in E. coli
ED3 gene fragments were amplified from RNA of TBEV Senzhang strain and digested with BamHI and XhoI. These were then ligated into a pET32a plasmid (Novagen, Hilton, Germany). The recombinant plasmid was used to transform E. coli BL21 (Novagen) cells to generate the ED3 antigen. Recombinant ED3 was verified by SDS-PAGE and purified using nickel affinity chromatography as described previously (Stenkova et al. 2017).
Expression of luciferase–ED3 fusion protein
A gene fragment of Nano luciferase (NLuc) was synthesized and inserted into the pcDNA3.1 plasmid. Then, the recombinant pcDNA3.1-NLuc plasmid was used as a vector to generate a NLuc–antigen fusion protein. Briefly, ED3 protein gene fragments were amplified and a stop codon was inserted directly after ED3 coding sequence by PCR. Then, the ED3 gene was digested by XbaI and NotI and then subcloned downstream of NLuc gene. The ED3 sequence of each plasmid construct was confirmed by PCR and DNA sequencing.
The recombinant pcDNA3.1-NLuc-ED3 plasmid was transfected into African Green Monkey SV40-transfected kidney fibroblast (COS7) cells to generate the recombinant protein antigen. Briefly, COS7 cells were seeded in a 24-well plate (at >85% confluence) and transfected using Lipofectamine® 2000 transfection reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. At 24, 48, 96 h post-transfection, NLuc activities in the supernatant containing the recombinant NLuc–ED3 fusion protein were detected separately using a luminometer (Promega, Madison, WI) after addition of a Coelenterazine substrate mix (Promega) (Issa et al. 2009). The supernatant was collected when the signal reached peak and then stored at −70°C until use.
Indirect ED3-ED3
One hundred microliters of purified ED3 antigen (TBEV ED3 antigen; 5 μg/mL) were coated onto wells of microtiter plates (Nunc, Germany) in 0.1 M carbonate buffer (pH 9.6) at 4°C overnight. After washing three times with phosphate-buffered saline (PBS), the wells were blocked by adding 200 μL bovine serum albumin (30 mg/mL in PBS) at 37°C for 1 h. After washing, 100 μL human serum, diluted 1:100 in PBS, was added. After incubation and washing, 100 μL of anti-human IgG peroxidase conjugate (Zhongshan, Inc., Beijing, China) was added (dilution 1:2000) and incubated at 37°C for 30 min. After washing, the assay was stained with tetramethyl-benzidine, stopped with 0.5 M H2SO4, and read at 450 nm.
Immunofluorescence assay
To perform IFA against TBEV, the IFA chips were prepared first as follows: BHK-21 cells infected with TBEV were incubated in the wells of glass chips at 37°C for 6 h, then the chips were washed with PBS and fixed with acetone for 1 h at −20°C. For IFA, the patient serum was diluted in 1:20 with PBS and added into the glass chip, 10 μL per well. After incubating at 37°C for 45 min, the chips were washed with PBS, and 20 μL of anti-human IgG FITC conjugate (Zhongshan, Inc.) was added into chip wells. After incubation and washing, the chips were visualized under fluorescence microscopy. Infected cells glowed light yellow to green if the patient sample was positive.
The NLuc-IC assay
The shematic of the NLuc-IC assay was shown in Figure 1. The NLuc-IC assay was performed at 37°C in an opaque 96-well plate. Briefly, sera were first diluted 1:10 in assay buffer A (20 mM Tris, pH 7.5, 150 mM NaCl, 5 mM MgCl2, and 1% Triton X-100). Next, 40 μL buffer A, 10 μL diluted serum (9 μL buffer A containing 1 μL serum), and 50 μL NLuc-ED3 (diluted in buffer A to yield 1 × 107 light units [LU]) were mixed together and added to each well for 1 h at 37°C. Next, 5 μL of a 30% suspension of Ultralink protein A/G Magnetic beads (Millipore) in PBS was added to each well of a 96-well plate. After 30 min at 37°C, the beads were washed four times with buffer A to remove unbounded antigen and antibody. After the final wash, NLuc substrate mix (Promega) was added to each well and LU intensity was measured in a Promega microplate luminometer (Berthold Technologies, Bad Wildbad, Germany) (Burbelo et al. 2009, Issa et al. 2009, Chidumayo et al. 2014). Data were expressed as mean LU from parallel triplet wells and corrected for background by subtracting the LU value of beads incubated with COS7 cell extracts in the absence of sera.

Schematic of the Luc-IC assay.
Statistical analysis
GraphPad Prism software (San Diego, CA) was used for statistical analysis. To calculate assay sensitivity and specificity, a cut-off limit for each antigen was set as the mean value of uninfected samples plus three standard deviations. One-way analysis of variance tests were used to compare the results for TBEV-uninfected cases and TBEV-infected cases. The level of significance was set at p < 0.05. Receiver operating characteristic (ROC) curves were also drawn to show the sensitivity and specificity of Nluc-IC and ED3-ELISA.
Results
Sensitivity and specificity of the TBEV NLuc-IC assay
We compared the results of NLuc-IC assay with those derived from ED3-ELISA and IFA. For specificity assay, we tested samples from 62 subjects who had no contact with TBEV or TBEV vaccines, including 10 samples from healthy persons and 52 samples from persons with a confirmed infection by another virus. Strong cross-reactions were observed in the IFA. Indeed, 10 of 15 samples from dengue fever patients, two of three samples from ZIKV-infected patients, 5 of 28 samples from JEV-infected patients, and 3 out of 3 samples from WNV-infected mice showed cross-reactions with the assay antigen. By contrast, neither NLuc-IC nor ED3-ELISA yielded false-positive results (Table 1).
Positive Cases Detected by Luc-IC, Immunofluorescence Assay, and ED3-ELISA
Samples included 50 clinical TBE cases, 28 JEV-infected cases, 3 ZIKV-infected cases, 3 sera from WNV-infected rabbits, sera from 15 clinical dengue cases, 3 sera from YFV-infected mice, and 10 sera from healthy people.
ED3, envelope domain III; ELISA, enzyme-linked immunosorbent assay; IFA, immunofluorescence assay; JEV, Japanese encephalitis virus; Luc-IC, luciferase-based immunocomplex assay system; NLuc, Nano luciferase; p/n, positive cases/total cases; TBEV, tick-borne encephalitis virus; WNV, West Nile virus; YFV, yellow fever virus; ZIKV, Zika virus.
For sensitivity assay, we tested sera from 50 clinically confirmed TBE cases. As shown in Table 1, IFA showed the greatest sensitivity: all 50 sera were positive. The NLuc-IC assay detected 47 positive samples (Fig. 2 and Table 1). By contrast, the ED3-ELISA showed the lowest sensitivity, detecting only 36 sera as positive. ROC curves were drawn to show the sensitivity and specificity of Nluc-IC and ED3-ELISA (Fig. 3).
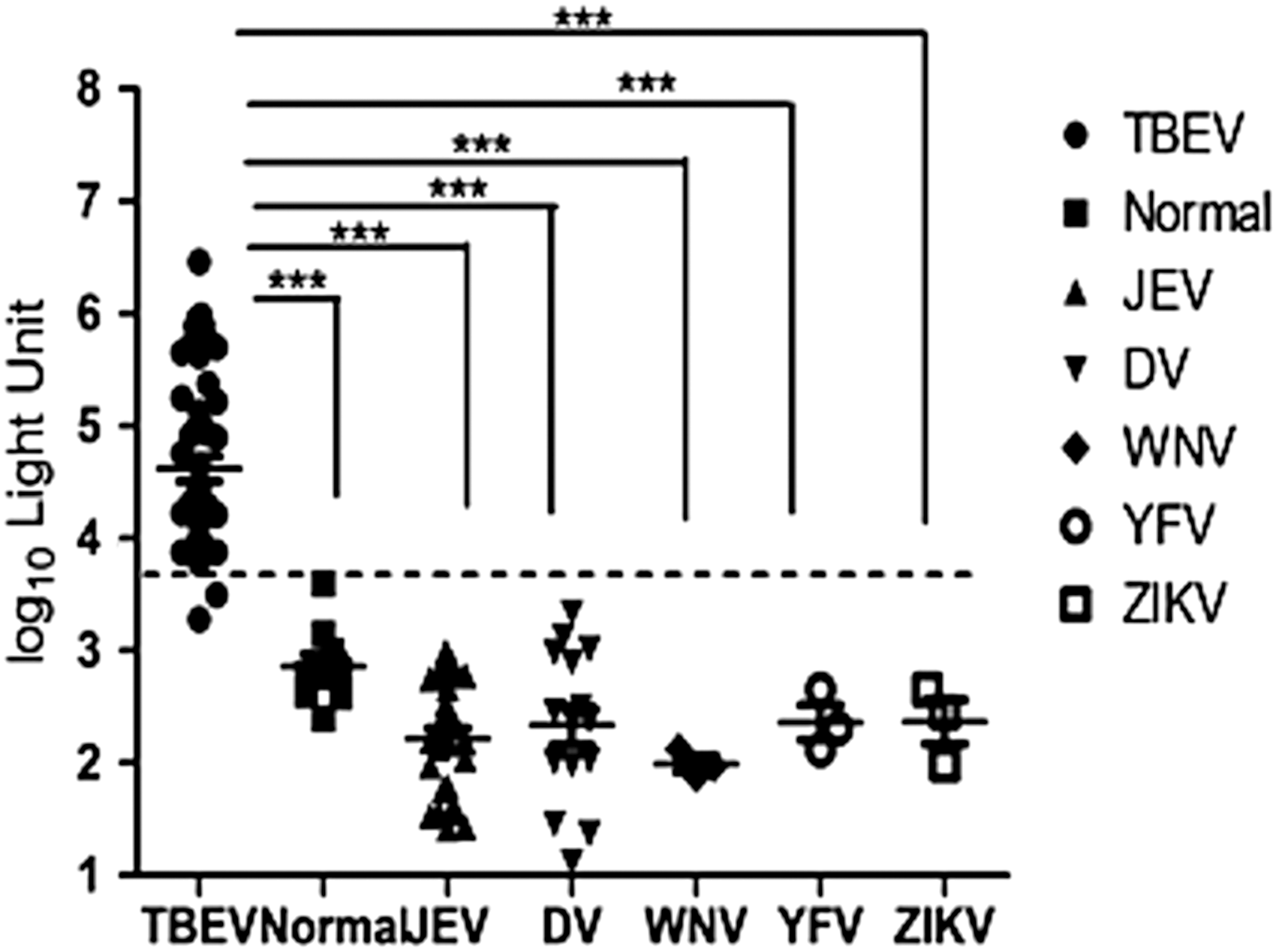
Detection of anti-TBEV antibodies in TBEV-, DV-, WNV-, YFV-, and ZIKV-infected samples and healthy controls using the Luc-IC assay. Each symbol represents the geometric mean for an individual serum sample (run in duplicate). The horizontal gray bars represent the median for each group. The dashed line represents the cut-off value for positivity in the NLuc-IC assay. NLuc, Nano luciferase; TBEV, tick-borne encephalitis virus; WNV, West Nile virus; YFV, yellow fever virus; ZIKV, Zika virus. ***p < 0.05.
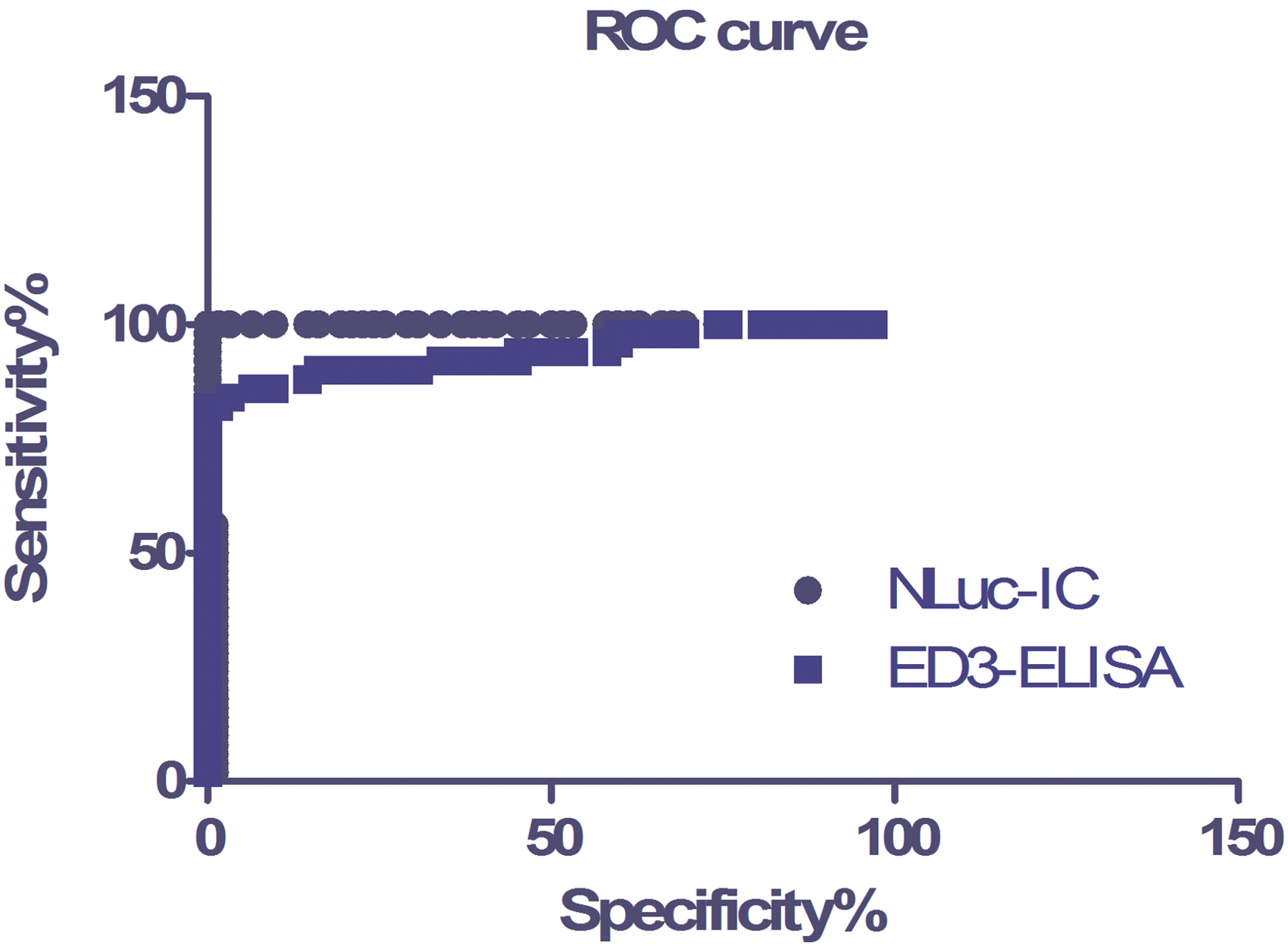
ROC curve of NLuc-IC and ED3-ELISA. Sensitivity and specificity comparison between NLuc-IC and ED3-ELISA by ROC analysis; AUC is 0.9990 for NLuc-IC and 0.9397 for ED3-ELISA. AUC, area under curve; ELISA, enzyme-linked immunosorbent assay; ROC, receiver operating characteristic. Color images are available online.
Because there is no gold standard serological assay for TBEV infection in Chinese hospitals, doctors often confirm TBE cases from the list of symptoms, epidemiological inquiry, and IFA results. However, because IFA shows a high level of cross-reactivity with other viruses, we re-examined the three samples detected positive by IFA but not by NLuc-IC.
To confirm the pathogen present in the three discordant samples, we tested them using commercial JEV and DENV ELISA kits. The results revealed that two samples were positive for JEV, and one was positive for DENV (Table 2); thus the three samples that tested positive in IFA but not NLuc-IC may have been false-positives. We tried to culture and isolate the virus from these samples to confirm the pathogen, but we were unsuccessful. Viral pathogens can be isolated from samples during the early stage of infection (Zhang et al. 2012), but these three patients had been infected for >2 weeks and were transferred from other hospitals. The treatments they received caused the vital titer to fall, which is why we could not isolate virus from the serum samples. We also performed a neutralizing test to identify the pathogen; the sera were unable to neutralize TBEV. However, serum from the two patients positive for JEV did neutralize JEV, and serum from the patient positive for DENV neutralized DENV; therefore, it is highly probable that these three sera were negative for TBEV.
Further Testing of the Three Cases That Yielded Different Results in the Luc-IC and Immunofluorescence Assays
DENV, dengue virus; N, negative; P, positive.
Discussion
Bioluminescence methods are very useful for detecting viral antigens due to their high sensitivity, broad dynamic range, and operational simplicity. Renilla luciferase is a sensitive reporter for a luciferase-immunoprecipitation system and has been used to detect antibodies to pathogens (Ludolfs et al. 2009, Peter et al. 2009). NLuc is a new kind of luciferase, which generates a glow-type luminescence (signal half-life >2 h) with an activity 150-fold greater than that of either firefly (Photinus pyralis) or renilla luciferase, which are similarly configured for glow-type assays.
To date, no immunoassay is based on NLuc. Here, we expressed a recombinant NLuc–ED3 fusion protein in COS7 cells and used it as the basis for an anti-TBEV antibody assay. The ED3 gene fragment was amplified from the far-east subtype, Senzhang strain. Though the ED3 sequence was highly variable among different flavivirus, the ED3 protein sequences were highly conserved for the three subtypes of TBEV, which is >90% sequence identity. So, this recombinant Nluc-ED3 would cover all of the three subtypes of TBEV infection.
The assay we developed was more specific for TBEV than the IFA, which identified three false-positive samples. The finding that these false-positives were most probably TBEV-negative suggests that the NLuc-IC assay was 100% sensitive and 100% specific for anti-TBEV antibodies.
The ED3 protein of TBEV is a specific antigen recognized by anti-TBEV antibodies (Holbrook et al. 2004, Niedrig et al. 2008, Chabierski et al. 2014, Stock et al. 2015). However, only a small fraction of anti-envelope antibodies target the ED3 region. Ludolfs et al. explored the use of an IC-ELISA to improve the sensitivity of ED3. They expressed a recombinant ED3 protein and labeled it with peroxidase. This was then incubated with serum, and the antibody–antigen complex was captured and detected. The results showed that the assay was 100% specific and 96% sensitive; indeed, it was much more sensitive than the ED3-ELISA (Ludolfs et al. 2009). The format of the NLuc-IC assay developed herein is similar to that of the IC-ELISA, which uses a labeled antigen to detect the antibody complex. However, developing the antigen for the new assay is much simpler than for the IC-ELISA. For the NLuc-IC assay, the antigen is fused to NLuc and expressed in a eukaryotic system; therefore, the labeled ED3 antigen can be collected directly from cell culture supernatant and used in the NLuc-IC assay; no antigen purification and labeling steps are necessary. Moreover, the recombinant NLuc-ED3 antigen expressed in eukaryotic cells is more similar to that of the native antigen (in terms of epitope conformation) than that produced in a prokaryotic system. Thus, the high specificity, high sensitivity, and simplicity of this NLuc-IC assay will be of great value in the clinic. In this assay, we used protein A/G-coupled beads to capture antigen–antibody complexes; thus both IgG and IgM antibodies are detected but cannot be distinguished. However, the NLuc-IC assay can be developed further to detect IgM or IgG using anti-IgM or anti-IgG protein-coupled beads to capture the antigen–antibody complexes. Thus, antibodies in acute phase or convalescent phase serum can be distinguished and identified.
Owing to the advantage of high accuracy and time saving, this NLuc-IC assay is a valuable tool for pathogen identification in laboratory. But it may not be suitable for clinical use right now, because of the need for expensive equipment reagent for NLuc activity detection. Meanwhile, the expressed amount of recombinant antigen in eucaryon system is limited, which may increase the cost of NLuc-IC assay. By validating this method for large-scale use and developing economic reagents and detector equipment, this NLuc-IC assay may be potentially useful both in laboratory and clinic use, both for humoral immune reaction surveillance and pathogen infection identification.
Footnotes
Acknowledgments
This work was supported by the National Natural Science Foundation of China (no. 81501789) and by the National Major Special Program of Science and Technology of China Foundation (2018ZX10302401, 2018ZX10101001-003 and AWS15J006).
Author Contributions
X.-P.K. contributed to the design of the study; Y.-C.L. and J.L. performed the experiments; X.-Y.W., Y.H., and N.-F.H. carried out data analysis and interpretation; X.-P.K. wrote the article. T.J. contributed to revise the article. All authors read and approved the final article.
Author Disclosure Statement
No conflicting financial interests exist.